











ZOLNOR HCT



Zolnor HCT 20 mg+5 mg+12,5 mg, comprimidos revestidos por película
Zolnor HCT 40 mg+5 mg+12,5 mg, comprimidos revestidos por película
Zolnor HCT 40 mg+10 mg+12,5 mg, comprimidos revestidos por película
Zolnor HCT 40 mg+5 mg+25 mg, comprimidos revestidos por película
Zolnor HCT 40 mg+10 mg+25 mg, comprimidos revestidos por película
Zolnor HCT 20 mg+5 mg+12,5 mg, comprimidos revestidos por película:
Cada comprimido revestido por película contém 20 mg de olmesartan medoxomilo, 5 mg de amlodipina (como besilato de amlodipina) e 12,5 mg de hidroclorotiazida.
Zolnor HCT 40 mg+5 mg+12,5 mg, comprimidos revestidos por película:
Cada comprimido revestido por película contém 40 mg de olmesartan medoxomilo, 5 mg de amlodipina (como besilato de amlodipina) e 12,5 mg de hidroclorotiazida.
Zolnor HCT 40 mg+10 mg+12,5 mg, comprimidos revestidos por película:
Cada comprimido revestido por película contém 40 mg de olmesartan medoxomilo, 10 mg de amlodipina (como besilato de amlodipina) e 12,5 mg de hidroclorotiazida.
Zolnor HCT 40 mg+5 mg+25 mg, comprimidos revestidos por película:
Cada comprimido revestido por película contém 40 mg de olmesartan medoxomilo, 5 mg de amlodipina (como besilato de amlodipina) e 25 mg de hidroclorotiazida.
Zolnor HCT 40 mg+10 mg+25 mg, comprimidos revestidos por película:
Cada comprimido revestido por película contém 40 mg de olmesartan medoxomilo, 10 mg de amlodipina (como besilato de amlodipina) e 25 mg de hidroclorotiazida.
Lista completa de excipientes, ver secção 6.1.
Comprimido revestido por película.
Zolnor HCT 20 mg+5 mg+12,5 mg, comprimidos revestidos por película:
Cor laranja clara, forma redonda, comprimido revestido por película de 8 mm e com C51 gravado num dos lados.
Zolnor HCT 40 mg+5 mg+12,5 mg, comprimidos revestidos por película:
Cor amarela clara, forma redonda, comprimido revestido por película de 9,5 mm e com C53 gravado num dos lados.
Zolnor HCT 40 mg+10 mg+12,5 mg, comprimidos revestidos por película:
Cor vermelha acinzentada, forma redonda, comprimido revestido por película de 9,5 mm e com C55 gravado num dos lados.
Zolnor HCT 40 mg+5 mg+25 mg, comprimidos revestidos por película:
Cor amarela clara, forma oval, comprimido revestido por película de 15 x 7 mm e com C54 gravado num dos lados.
Zolnor HCT 40 mg+10 mg+25 mg, comprimidos revestidos por película:
Cor vermelha acinzentada, forma oval, comprimido revestido por película de 15 x 7 mm e com C57 gravado num dos lados.
Tratamento da hipertensão essencial.
Terapêutica adjuvante
Zolnor HCT está indicado em doentes adultos cuja tensão arterial não está adequadamente controlada com a combinação de olmesartan medoxomilo e amlodipina tomados através de uma formulação de dois componentes.
Terapêutica de substituição
Zolnor HCT está indicado como terapêutica de substituição em doentes adultos cuja tensão arterial está adequadamente controlada com a combinação de olmesartan medoxomilo, amlodipina e hidroclorotiazida, tomados através de uma formulação com dois componentes (olmesartan medoxomilo e amlodipina ou olmesartan medoxomilo e hidroclorotiazida) e uma formulação de um componente (hidroclorotiazida ou amlodipina).
Posologia
Adultos
A dose recomendada de Zolnor HCT é de um comprimido por dia.
Terapêutica adjuvante
Zolnor HCT 20 mg+5 mg+12,5 mg pode ser administrado em doentes cuja tensão arterial não está adequadamente controlada com olmesartan medoxomilo 20 mg e amlodipina 5 mg tomados através de uma formulação de dois componentes.
Zolnor HCT 40 mg+5 mg+12,5 mg pode ser administrado em doentes cuja tensão arterial não está adequadamente controlada com olmesartan medoxomilo 40 mg e amlodipina 5 mg tomados através de uma formulação de dois componentes ou em doentes cuja tensão arterial não está adequadamente controlada com Zolnor HCT 20 mg+5 mg+12,5 mg.
Zolnor HCT 40 mg+5 mg+25 mg pode ser administrado em doentes cuja tensão arterial não está adequadamente controlada com Zolnor HCT 40 mg+5 mg+12,5 mg.
Zolnor HCT 40 mg+10 mg+12,5 mg pode ser administrado em doentes cuja tensão arterial não está adequadamente controlada com olmesartan medoxomilo 40 mg e amlodipina 10 mg tomados através de uma formulação de dois componentes ou com Zolnor HCT 40 mg+5 mg+12,5 mg.
Zolnor HCT 40 mg+10 mg+25 mg pode ser administrado em doentes cuja tensão arterial não está adequadamente controlada com Zolnor HCT 40 mg+10 mg+12,5 mg ou com Zolnor HCT 40 mg+5 mg+25 mg.
Recomenda-se a titulação gradual da dose dos componentes individuais antes da mudança para a combinação tripla. Quando clinicamente apropriado pode ser considerada uma mudança direta da formulação de dois componentes para a combinação tripla.
Terapêutica de substituição
Os doentes controlados com doses estáveis de olmesartan medoxomilo, amlodipina e hidroclorotiazida tomados ao mesmo tempo, através de uma formulação com dois componentes (olmesartan medoxomilo e amlodipina ou olmesartan medoxomilo e hidroclorotiazida) e uma formulação de um componente (hidroclorotiazida ou amlodipina) podem mudar para Zolnor HCT contendo os componentes nas mesmas doses.
A dose máxima recomendada de Zolnor HCT é de 40 mg+10 mg+ 25 mg por dia.
Idosos (idade igual ou superior a 65 anos)
Em doentes idosos é recomendada precaução, incluindo uma monitorização mais frequente da tensão arterial, particularmente com a dose máxima de Zolnor HCT de 40 mg+10 mg+25 mg por dia.
Um aumento de dosagem em doentes idosos deve ser efetuado com precaução (ver secções 4.4 e 5.2).
Estão disponíveis dados muito limitados sobre o uso de Zolnor HCT em doentes com idade igual ou superior a 75 anos. Recomenda-se extrema precaução, incluindo uma monitorização mais frequente da tensão arterial.
Compromisso renal
A dose máxima em doentes com compromisso renal ligeiro a moderado (depuração da creatinina de 30-60 ml/min) é Zolnor HCT 20 mg+5 mg+12,5 mg, devido à experiência limitada com a dose de 40 mg de olmesartan medoxomilo neste grupo de doentes.
É aconselhável a monitorização das concentrações séricas de potássio e de creatinina em doentes com compromisso renal moderado.
A utilização de Zolnor HCT em doentes com compromisso renal grave (depuração da creatinina < 30 ml/min) está contraindicada (ver secções 4.3, 4.4 e 5.2).
Compromisso hepático
Zolnor HCT deve ser usado com precaução em doentes com compromisso hepático ligeiro (ver secções 4.4 e 5.2).
Em doentes com compromisso hepático moderado, a dose máxima não deve exceder Zolnor HCT 20 mg+5 mg+12,5 mg uma vez por dia. É aconselhável uma monitorização cuidadosa da tensão arterial e da função renal em doentes com compromisso hepático.
Como acontece com todos os antagonistas dos canais de cálcio, a semivida da amlodipina é prolongada em doentes com compromisso hepático, não tendo sido estabelecidas recomendações posológicas. Consequentemente, Zolnor HCT deve ser administrado com precaução nestes doentes. A farmacocinética da amlodipina não foi estudada no compromisso hepático grave. Em doentes com compromisso hepático, a amlodipina deve ser iniciada na dose mais baixa e titulada lentamente.
A utilização de Zolnor HCT está contraindicada em doentes com compromisso hepático grave (ver secções 4.3 e 5.2), colestase ou obstrução biliar (ver secção 4.3).
População pediátrica
Zolnor HCT não é recomendado em doentes com idade inferior a 18 anos devido à ausência de dados de segurança e eficácia.
Modo de administração:
O comprimido deve ser engolido com uma quantidade suficiente de líquido (por exemplo, um copo de água). O comprimido não deve ser mastigado e deve ser tomado à mesma hora todos os dias.
Zolnor HCT pode ser tomado com ou sem alimentos.
Hipersensibilidade às substâncias ativas, aos derivados da di-hidropiridina ou a substâncias derivadas da sulfonamida (dado que a hidroclorotiazida é um fármaco derivado da sulfonamida), ou a qualquer um dos excipientes mencionados na secção 6.1.
Compromisso renal grave (ver secções 4.4 e 5.2).
Hipocaliemia refractária, hipercalcemia, hiponatremia e hiperuricemia sintomática.
Insuficiência hepática grave, colestase e afeções biliares obstrutivas (ver secção 5.2).
Segundo e terceiro trimestres de gravidez (ver secções 4.4 e 4.6).
O uso concomitante de Zolnor HCT com medicamentos contendo aliscireno é contraindicado em doentes com diabetes mellitus ou compromisso renal (TFG < 60 ml/min./1,73 m2) (ver secções 4.5 e 5.1).
Devido ao componente amlodipina, Zolnor HCT está contraindicado em doentes com:
- Choque (incluindo choque cardiogénico).
- Hipotensão grave.
- Obstrução do trato de saída do ventrículo esquerdo (por exemplo, estenose aórtica de grau elevado).
- Insuficiência cardíaca hemodinamicamente instável após um enfarte agudo do miocárdio.
Doentes com hipovolémia ou depleção de sódio:
Pode ocorrer hipotensão sintomática, especialmente após a primeira toma, em doentes com depleção de volume e/ou de sódio como resultado da terapêutica diurética intensiva, restrição de sal na dieta, diarreia ou vómitos. Recomenda-se a correção desta situação antes da administração de Zolnor HCT ou supervisão médica cuidadosa no início do tratamento.
Outras afeções que estimulam o sistema renina-angiotensina-aldosterona:
Em doentes cujo tónus vascular e função renal dependem predominantemente da atividade do sistema renina-angiotensina-aldosterona (por exemplo, doentes com insuficiência cardíaca congestiva grave ou com doença renal subjacente, incluindo estenose da artéria renal), o tratamento com medicamentos que afetam este sistema foi associado a hipotensão aguda, azotemia, oligúria ou, raramente, a insuficiência renal aguda.
Hipertensão renovascular:
Existe um risco acrescido de hipotensão grave e de insuficiência renal quando doentes com estenose bilateral das artérias renais ou estenose da artéria que irriga um rim único funcionante são tratados com medicamentos que afetam o sistema renina-angiotensina-aldosterona.
Compromisso renal e transplante renal:
Recomenda-se a monitorização periódica das concentrações séricas de potássio e creatinina quando Zolnor HCT é utilizado em doentes com compromisso renal.
Não é recomendada a utilização de Zolnor HCT em doentes com compromisso renal grave (depuração da creatinina < 30 ml/min) (ver secções 4.2, 4.3 e 5.2).
Em doentes com compromisso renal pode ocorrer azotemia associada aos diuréticos tiazídicos.
Se se verificar que o grau de compromisso renal aumenta, é necessária a reavaliação cuidadosa do tratamento, podendo considerar-se a possibilidade de suspensão da terapêutica diurética.
Não existe experiência com a administração de Zolnor HCT em doentes submetidos a transplante renal recente ou em doentes com uma disfunção renal terminal (isto é, depuração da creatinina < 12 ml/min).
Duplo bloqueio do sistema renina-angiotensina-aldosterona (SRAA):
Existe evidência de que o uso concomitante de inibidores da ECA, antagonistas dos recetores da angiotensina II ou aliscireno aumenta o risco de hipotensão, hipercaliemia e função renal diminuída (incluindo insuficiência renal aguda). O duplo bloqueio do SRAA através do uso combinado de inibidores da ECA, antagonistas dos recetores da angiotensina II ou aliscireno, é portanto, não recomendado (ver secções 4.5 e 5.1).
Se a terapêutica de duplo bloqueio for considerada absolutamente necessária, esta só deverá ser utilizada sob a supervisão de um especialista e sujeita a uma monitorização frequente e apertada da função renal, eletrólitos e pressão arterial.
Os inibidores da ECA e os antagonistas dos recetores da angiotensina II não devem ser utilizados concomitantemente em doentes com nefropatia diabética.
Compromisso hepático:
A exposição à amlodipina e ao olmesartan medoxomilo é maior em doentes com compromisso hepático (ver secção 5.2).
Além disso, pequenas alterações no equilíbrio hidroeletrolítico durante a terapêutica tiazídica podem precipitar coma hepático em doentes com compromisso hepático ou doença hepática progressiva.
Deve ter-se precaução ao administrar Zolnor HCT a doentes com compromisso hepático ligeiro a moderado.
Em doentes com compromisso hepático moderado, a dose de olmesartan medoxomilo não deve exceder 20 mg (ver secção 4.2).
Em doentes com compromisso hepático, a amlodipina deve ser iniciada na dose mais baixa do intervalo de dosagens e deve ser tomada precaução no início do tratamento e quando se aumenta a dose.
A utilização de Zolnor HCT está contraindicada em doentes com compromisso hepático grave, colestase ou obstrução biliar (ver secção 4.3).
Estenose da válvula aórtica ou mitral, cardiomiopatia hipertrófica obstrutiva:
Devido ao componente amlodipina de Zolnor HCT, como acontece com outros vasodilatadores, recomenda-se precaução especial em doentes com estenose aórtica ou mitral ou cardiomiopatia hipertrófica obstrutiva.
Aldosteronismo primário:
Geralmente, os doentes com aldosteronismo primário não respondem aos fármacos anti-hipertensores que atuam através da inibição do sistema renina-angiotensina. Por conseguinte, não se recomenda a utilização de Zolnor HCT nestes doentes.
Efeitos metabólicos e endócrinos:
A terapêutica com tiazidas pode diminuir a tolerância à glucose. Em doentes diabéticos poderá ser necessário ajuste posológico de insulina ou de agentes hipoglicemiantes orais (ver secção 4.5). Durante a terapêutica tiazídica a diabetes mellitus latente pode manifestar-se.
Níveis de colesterol e triglicéridos aumentados são efeitos indesejáveis conhecidos associados à terapêutica diurética tiazídica.
Em alguns doentes sob terapêutica com tiazidas pode ocorrer hiperuricemia ou desencadear-se uma crise de gota.
Desequilíbrio eletrolítico:
Tal como em qualquer doente sob terapêutica diurética, deverá ser feita a determinação periódica dos eletrólitos séricos em intervalos adequados.
As tiazidas, incluindo a hidroclorotiazida, podem causar desequilíbrio hidroeletrolítico (incluindo hipocaliemia, hiponatremia e alcalose hipoclorémica). Os sinais de alerta de desequilíbrio hidroeletrolítico são secura da boca, sede, fraqueza, letargia, sonolência, agitação, dores musculares ou cãibras, fadiga muscular, hipotensão, oligúria, taquicardia e distúrbios gastrointestinais tais como náuseas e vómitos (ver secção 4.8).
O risco de hipocaliemia é maior em doentes com cirrose hepática, em doentes com estimulação da diurese, em doentes que recebem uma quantidade inadequada de eletrólitos por via oral e em doentes sob terapêutica concomitante com corticosteroides ou ACTH (ver secção 4.5).
Por outro lado, devido ao antagonismo dos recetores de angiotensina II (AT1) pelo componente olmesartan medoxomilo do Zolnor HCT, pode ocorrer hipercaliemia, especialmente em presença de compromisso renal e/ou de insuficiência cardíaca e de diabetes mellitus. Recomenda-se uma monitorização cuidadosa do potássio sérico em doentes de risco. Os diuréticos poupadores de potássio, os suplementos de potássio ou os substitutos do sal contendo potássio e outros medicamentos que possam elevar os níveis séricos de potássio (por exemplo, a heparina) devem ser administrados concomitantemente com precaução com Zolnor HCT (ver secção 4.5) e com monitorização frequente dos níveis de potássio.
Não existe evidência de que o olmesartan medoxomilo possa reduzir ou prevenir a hiponatremia induzida por diuréticos. O défice de cloreto é geralmente ligeiro e habitualmente não requer tratamento.
As tiazidas podem diminuir a excreção urinária do cálcio e causar um aumento ligeiro e intermitente do cálcio sérico na ausência de perturbações conhecidas do metabolismo do cálcio. A hipercalcemia pode evidenciar um hiperparatiroidismo não diagnosticado. As tiazidas deverão ser suspensas antes de se efetuarem testes da função paratiroideia.
Demonstrou-se que as tiazidas aumentam a excreção urinária de magnésio, o que pode resultar em hipomagnesemia.
Com temperaturas elevadas pode ocorrer hiponatremia de diluição em doentes com edemas.
Lítio:
Como com outros antagonistas dos recetores da angiotensina II, não é recomendada a administração concomitante de Zolnor HCT e lítio (ver secção 4.5).
Insuficiência cardíaca:
Em consequência da inibição do sistema renina-angiotensina-aldosterona, podem ser esperadas alterações na função renal em indivíduos suscetíveis.
Em doentes com insuficiência cardíaca grave, cuja função renal possa depender da atividade do sistema renina-angiotensina-aldosterona, o tratamento com inibidores da enzima de conversão da angiotensina (ECA) e antagonistas dos recetores da angiotensina tem sido associado a oligúria e/ou azotemia progressiva e (raramente) a insuficiência renal aguda e/ou morte.
Os doentes com insuficiência cardíaca devem ser tratados com precaução. Num estudo de longa duração com amlodipina, controlado com placebo, em doentes com insuficiência cardíaca grave (classes III e IV da NYHA), a incidência descrita de edema pulmonar foi maior no grupo tratado com amlodipina do que no grupo com placebo (ver secção 5.1). Os bloqueadores dos canais de cálcio, incluindo a amlodipina, devem ser utilizados com precaução em doentes com insuficiência cardíaca congestiva, pois podem aumentar o risco de eventos cardiovasculares futuros e a mortalidade.
Enteropatia semelhante a esprue:
Em casos muito raros foi notificada diarreia crónica grave com perda de peso substancial em doentes a tomar olmesartan, alguns meses a anos após o início do medicamento, possivelmente causada por uma reação de hipersensibilidade retardada localizada. As biópsias intestinais dos doentes demonstraram muitas vezes atrofia das vilosidades. Se um doente desenvolver estes sintomas durante o tratamento com olmesartan, e na ausência de outras etiologias aparentes, o tratamento com olmesartan deve ser imediatamente descontinuado e não deve ser reiniciado. Se a diarreia não melhorar durante a semana após a descontinuação da terapêutica, deve ser considerada a opinião de um especialista (por exemplo um gastroenterologista).
Efusão coroidal, miopia Aguda e Glaucoma Agudo de Ângulo Fechado Secundário:
A hidroclorotiazida, uma sulfonamida, pode causar uma reação idiossincrática, resultando em efusão coroidal com perda do campo visual, miopia aguda transitória e glaucoma agudo de ângulo fechado. Os sintomas incluem início agudo da diminuição da acuidade visual ou dor ocular que geralmente ocorrem dentro de horas a semanas, após o início do medicamento. O glaucoma agudo de ângulo fechado, não tratado, pode levar à perda permanente da visão. O tratamento primário consiste em interromper a hidroclorotiazida, tão rapidamente quanto possível. Podem ter que ser considerados tratamentos médicos ou cirúrgicos imediatos, caso a pressão intra-ocular permaneça não controlada. Os fatores de risco para o desenvolvimento de glaucoma agudo de ângulo fechado pode incluir história de alergia à sulfonamida ou penicilina (Ver secção 4.8).
Gravidez:
Os antagonistas da angiotensina II (ARA) não devem ser iniciados durante a gravidez. A não ser em situações em que a manutenção da terapêutica com ARA seja considerada essencial, nas doentes que planeiem engravidar o tratamento deve ser alterado para anti-hipertensores cujo perfil de segurança durante a gravidez esteja estabelecido. Quando é diagnosticada a gravidez, o tratamento com ARA deve ser interrompido imediatamente e, se apropriado, deverá ser iniciada uma terapêutica alternativa (ver secções 4.3 e 4.6).
População pediátrica:
Zolnor HCT não está indicado em crianças e adolescentes com idade inferior a 18 anos.
Doentes idosos:
Em idosos, o aumento da dosagem deve ser efetuado com precaução (ver secção 5.2).
Fotossensibilidade:
Foram relatados casos de reações de fotossensibilidade com diuréticos tiazídicos (ver secção 4.8). Se a reação de fotossensibilidade ocorrer durante o tratamento com Zolnor HCT, recomenda-se a interrupção do tratamento. Se for considerado necessário retomar a administração do diurético, recomenda-se a proteção das áreas expostas ao sol ou à radiação UVA artificial.
Cancro da pele não-melanoma
Em dois estudos epidemiológicos baseados no registo nacional de cancro da Dinamarca foi observado um aumento do risco de cancro da pele não-melanoma (NMSC) [carcinoma basocelular (BCC) e carcinoma espinocelular (SCC)] com uma dose cumulativa crescente de exposição a hidroclorotiazida (HCTZ). A atividade fotossensibilizadora da HCTZ pode atuar como mecanismo para o MNSC.
Os doentes em tratamento com HCTZ devem ser informados do risco de NMSC e aconselhados a observar regularmente a sua pele. Quaisquer novas lesões da pele suspeitas devem ser imediatamente comunicadas ao médico. Os doentes devem ser aconselhados a tomar medidas preventivas tais como limitação da exposição à luz solar e à radiação ultravioleta e, em caso de exposição, a utilização de proteção adequada com vista a mininizar o risco de cancro da pele. As lesões cutâneas suspeitas devem ser rapidamente examinadas, nomeadamente através de exames histológicos de biópsias. A utilização de HCTZ também poderá ter que ser reavaliada em doentes com antecedentes de NMSC (ver também secção 4.8).
Toxicidade respiratória aguda
Foram notificados casos muito raros graves de toxicidade respiratória aguda, incluindo síndrome da insuficiência respiratória aguda (ARDS), após a toma de hidroclorotiazida. O edema pulmonar desenvolve-se tipicamente no espaço de minutos ou horas após a toma de hidroclorotiazida. No início, os sintomas incluem dispneia, febre, deterioração pulmonar e hipotensão. Em caso de suspeita de diagnóstico de ARDS, Zolnor HCT deve ser retirado e deve ser administrado o tratamento adequado. A hidroclorotiazida não deve ser administrada a doentes que tenham apresentado anteriormente ARDS após a toma de hidroclorotiazida.
Outras:
Como acontece com outros agentes anti-hipertensores, a diminuição excessiva da tensão arterial em doentes com doença cardíaca isquémica ou com doença vascular cerebral isquémica pode resultar em enfarte do miocárdio ou em acidente vascular cerebral.
Podem ocorrer reações de hipersensibilidade à hidroclorotiazida em doentes com ou sem antecedentes de alergia ou asma brônquica, embora sejam mais prováveis em doentes com estes antecedentes.
Foi referida a exacerbação ou ativação de lúpus eritematoso sistémico com a utilização de diuréticos tiazídicos.
Como com todos os outros antagonistas dos recetores da angiotensina II, o efeito de diminuição da tensão arterial do olmesartan é ligeiramente menor em doentes de raça negra do que em doentes de outras raças. No entanto, este efeito não foi observado em um dos três ensaios clínicos com Zolnor HCT que incluiu doentes de raça negra (30%), ver também a secção 5.1.
Interações potenciais relacionadas com a combinação Zolnor HCT: Uso concomitante não recomendado
Lítio:
Foram notificados aumentos reversíveis das concentrações séricas e da toxicidade do lítio durante a administração concomitante de lítio com inibidores da enzima de conversão da angiotensina e, raramente, com antagonistas dos recetores da angiotensina II. Além disso, a depuração renal do lítio é reduzida pelas tiazidas e consequentemente pode aumentar o risco de toxicidade por lítio. Por conseguinte, não é recomendada a utilização concomitante de Zolnor HCT e lítio (ver secção 4.4). Se a utilização concomitante for necessária recomenda-se uma monitorização cuidadosa dos níveis séricos de lítio.
Uso concomitante que requer precaução
Baclofeno:
Pode ocorrer uma potenciação do efeito anti-hipertensor.
Medicamentos anti-inflamatórios não esteroides:
AINEs (i.e. ácido acetilsalicílico (>3 g/dia), inibidores da COX-2 e AINEs não seletivos) podem reduzir o efeito anti-hipertensor dos diuréticos tiazídicos e dos antagonistas dos recetores da angiotensina II.
Em alguns doentes com a função renal comprometida (por exemplo, doentes desidratados ou doentes idosos com função renal comprometida), a administração concomitante de antagonistas dos recetores da angiotensina II e de agentes inibidores da cicloxigenase pode resultar numa deterioração adicional da função renal, incluindo possível insuficiência renal aguda, geralmente reversível. Por conseguinte, a combinação deve ser administrada com precaução, especialmente nos idosos. Os doentes devem ser adequadamente hidratados e deve ser dada particular atenção à monitorização da função renal no início da terapêutica concomitante e regularmente ao longo do tratamento.
Uso concomitante a ter em consideração Amifostina:
Pode ocorrer uma potenciação do efeito anti-hipertensor.
Outros agentes anti-hipertensores:
O efeito de redução da tensão arterial do Zolnor HCT pode ser potenciado pela utilização concomitante de outros fármacos anti-hipertensores.
Álcool, barbitúricos, narcóticos ou antidepressivos:
Pode ocorrer uma potenciação da hipotensão ortostática. Interações potenciais relacionadas com olmesartan medoxomilo:
Uso concomitante não recomendado
Inibidores da ECA, antagonistas dos recetores da angiotensina II ou aliscireno:
Os dados de ensaios clínicos têm demonstrado que o duplo bloqueio do sistema renina-angiotensina-aldosterona (SRAA) através do uso combinado de inibidores da ECA, antagonistas dos recetores da angiotensina II ou aliscireno está associado a uma maior frequência de acontecimentos adversos, tais como hipotensão, hipercaliemia e função renal diminuída (incluindo insuficiência renal aguda) em comparação com o uso de um único fármaco com ação no SRAA (ver secções 4.3, 4.4 e 5.1).
Medicamentos que alteram os níveis de potássio:
A utilização concomitante de diuréticos poupadores de potássio, de suplementos de potássio, de substitutos do sal que contenham potássio ou de outros medicamentos que possam aumentar os níveis séricos de potássio (por exemplo, heparina inibidores da ECA) pode causar aumento do potássio sérico (ver secção 4.4). Se forem prescritos concomitantemente com Zolnor HCT medicamentos que afetam o potássio, recomenda-se uma monitorização do potássio sérico.
Informação adicional
Agente sequestrador de ácidos biliares, colessevelam:
A administração concomitante do agente sequestrador de ácidos biliares, cloridrato de colessevelam, reduz a exposição sistémica e o pico de concentração plasmática do olmesartan e reduz o t1/2. A administração de olmesartan medoxomilo pelo menos 4 horas antes da administração de cloridrato de colessevelam diminuiu o efeito de interação dos fármacos. Deve considerar-se administrar o olmesartam medoxomilo pelo menos 4 horas antes da administração de cloridrato de colessevelam (ver secção 5.2).
Após tratamento com antiácidos (hidróxido de alumínio e magnésio) registou-se uma ligeira redução na biodisponibilidade do olmesartan.
O olmesartan medoxomilo não teve efeito significativo na farmacocinética ou na farmacodinâmica da varfarina nem na farmacocinética da digoxina.
A administração concomitante de olmesartan medoxomilo com pravastatina não teve efeitos clinicamente relevantes na farmacocinética de cada componente em indivíduos saudáveis.
O olmesartan não teve efeitos inibidores clinicamente relevantes nas enzimas 1A1/2, 2A6, 2C8/9, 2C19, 2D6, 2E1 e 3A4 do citocromo P450 humano in vitro, e não teve efeitos ou teve efeitos indutores mínimos nas atividades do citocromo P450 do rato. Não são esperadas interações clinicamente relevantes entre o olmesartan e medicamentos metabolizados pelas enzimas do citocromo P450 acima mencionadas.
Interações potenciais relacionadas com amlodipina Uso concomitante que requer precaução
Efeitos de outros medicamentos na amlodipina Inibidores do CYP3A4:
A utilização concomitante da amlodipina com inibidores fortes ou moderados do CYP3A4 (inibidores das proteases, antifúngicos azóis, macrólidos como a eritromicina ou a claritromicina, verapamilo ou diltiazem) pode conduzir a um aumento significativo da exposição à amlodipina. A tradução clínica destas variações farmacocinéticas pode ser mais pronunciada nos idosos. Poderão assim ser necessários monitorização clínica e ajustamento da dose.
Indutores do CYP3A4:
Não existem dados disponíveis sobre o efeito dos indutores do CYP3A4 na amlodipina. A utilização concomitante de indutores do CYP3A4 (por exemplo, rifampicina, Hypericum perforatum [hipericão]) pode originar a uma concentração plasmática mais baixa de amlodipina. A amlodipina deve ser utilizada com precaução juntamente com indutores do CYP3A4.
Não é recomendada a administração de amlodipina com toranja ou sumo de toranja, pois em alguns doentes a biodisponibilidade pode aumentar resultando num aumento do efeito de diminuição da tensão arterial.
Dantroleno (perfusão): Em animais, foram observadas fibrilhação ventricular letal e colapso cardiovascular em associação com hipercaliemia após administração de verapamilo e dantroleno intravenoso. Devido ao risco de hipercaliemia, recomenda- se que a administração concomitante de bloqueadores de canais de cálcio, como a amlodipina, seja evitada em doentes suscetíveis a hipertermia maligna e no controlo da hipertermia maligna.
Efeitos da amlodipina noutros medicamentos
O efeito de diminuição da tensão arterial da amlodipina adiciona-se aos efeitos de diminuição da tensão arterial de outros agentes anti-hipertensores.
Em estudos clínicos de interação, a amlodipina não afetou a farmacocinética da atorvastatina, digoxina ou varfarina.
Sinvastatina: A administração concomitante de doses múltiplas de 10 mg de amlodipina com 80 mg de sinvastatina resultou num aumento de 77% na exposição à sinvastatina comparativamente com a sinvastatina isolada. Nos doentes a tomar amlodipina deve limitar-se a dose de sinvastatina a 20 mg por dia.
Tacrolímus: Quando administrado concomitantemente com amlodipina, existe um risco de aumento dos níveis sanguíneos de tacrolímus. De forma a evitar a toxicidade do tacrolimus, a administração de amlodipina em doentes tratados com tacrolímus necessita de uma monitorização dos níveis sanguíneos de tacrolímus e um ajuste na dose de tacrolímus, sempre que necessário.
Inibidores Mecânicos do Alvo da Rapamicina (mTOR): inibidores da mTOR, como sirolimus, temsirolimus e everolimus, são substratos do CYP3A. A amlodipina é um inibidor fraco do CYP3A. Com a utilização concomitante de inibidores da mTOR, a amlodipina pode aumentar a exposição dos inibidores da mTOR.
Ciclosporina: Num estudo prospetivo em doentes submetidos a transplante renal, observou-se um aumento médio de 40% nos níveis mínimos de ciclosporina quando usado concomitantemente com a amlodipina. A administração concomitante de Zolnor HCT com ciclosporina pode aumentar a exposição à ciclosporina. Deve ser considerada a monitorização dos níveis mínimos de ciclosporina durante a utilização concomitante e a redução das doses de ciclosporina, conforme necessário.
Interações potenciais relacionadas com hidroclorotiazida: Uso concomitante não recomendado
Medicamentos que alteram os níveis de potássio:
O efeito depletor de potássio da hidroclorotiazida (ver secção 4.4) pode ser potenciado pela administração concomitante de outros fármacos associados à diminuição de potássio e hipocaliemia (por exemplo, outros diuréticos caliuréticos, laxantes, corticosteroides, ACTH, anfotericina, carbenoxolona, penicilina G sódica ou derivados de ácido salicílico). Não se recomenda, portanto, o uso concomitante destes fármacos.
Uso concomitante que requer precaução Sais de cálcio:
Os diuréticos tiazídicos podem aumentar o cálcio sérico devido à diminuição da sua excreção. Se a prescrição de suplementos de cálcio for necessária, o cálcio sérico deve ser monitorizado e a dosagem do cálcio deve ser ajustada em conformidade.
Colestiramina e resinas de colestipol:
A absorção da hidroclorotiazida é prejudicada em presença de resinas de troca aniónica.
Glicosidos digitálicos:
A hipocaliemia ou a hipomagnesemia induzida por diuréticos tiazídicos podem favorecer o início de arritmias cardíacas induzidas por digitálicos.
Medicamentos afetados pelos desequilíbrios do potássio sérico:
É recomendada uma monitorização periódica do potássio sérico e a realização de ECG quando o Zolnor HCT é administrado com medicamentos afetados pelos desequilíbrios do potássio sérico (por exemplo, glicosidos digitálicos e antiarrítmicos) e com os seguintes medicamentos indutores de “torsades de pointes" (taquicardia ventricular) (incluindo alguns antiarrítmicos), sendo a hipocaliemia um fator de predisposição para “torsades de pointes" (taquicardia ventricular):
- Antiarrítmicos Classe Ia (por exemplo, quinidina, hidroquinidina, disopiramida).
- Antiarrítmicos Classe III (por exemplo, amiodarona, sotalol, dofetilida, ibutilida).
- Alguns antipsicóticos (por exemplo, tioridazina, cloropromazina, levomepromazina, trifluoperazina, ciamemazina, sulpirida, sultoprida, amissulprida, tiaprida, pimozida, haloperidol, droperidol)
- Outros (por exemplo, bepridilo, cisaprida, difemanil, eritromicina I.V., halofantrina, mizolastina, pentamidina, esparfloxacina, terfenadina, vincamina I.V.).
Relaxantes não despolarizantes do músculoesquelético (por exemplo, tubocurarina): O efeito dos relaxantes não despolarizantes do músculoesquelético pode ser potenciado pela hidroclorotiazida.
Agentes anticolinérgicos (por exemplo, atropina, biperideno):
Aumento da biodisponibilidade dos diuréticos tiazídicos pela diminuição da motilidade gastrointestinal e do ritmo de esvaziamento do estômago.
Medicamentos antidiabéticos (agentes orais e insulina):
O tratamento com tiazida pode influenciar a tolerância à glucose. Pode ser necessário ajustar a dosagem do medicamento antidiabético (ver secção 4.4).
Metformina:
A metformina deve ser utilizada com precaução devido ao risco de acidose láctica induzida por uma possível insuficiência renal funcional relacionada com a hidroclorotiazida.
Bloqueadores beta e diazóxido:
O efeito hiperglicémico dos bloqueadores beta e do diazóxido pode ser potenciado pelas tiazidas.
Aminas vasopressoras (por exemplo, noradrenalina): O efeito das aminas vasopressoras pode ser diminuído.
Medicamentos utilizados no tratamento da gota (por exemplo, probenecida, sulfimpirazona e alopurinol):
Poderá ser necessário proceder a um ajuste de dose dos fármacos uricosúricos uma vez que a hidroclorotiazida pode aumentar os níveis séricos de ácido úrico. Poderá ser necessário aumentar a dosagem de probenecida ou sulfimpirazona. A administração concomitante de tiazídicos pode aumentar a incidência de reações de hipersensibilidade ao alopurinol.
Amantadina:
As tiazidas podem aumentar o risco de efeitos adversos causados pela amantadina.
Agentes citotóxicos (por exemplo, ciclofosfamida, metotrexato):
As tiazidas podem diminuir a excreção renal de medicamentos citotóxicos e potenciar os seus efeitos mielossupressores.
Salicilatos:
No caso de dosagens elevadas de salicilatos, a hidroclorotiazida pode aumentar o efeito tóxico dos salicilatos no sistema nervoso central.
Metildopa:
Existem notificações isoladas de anemia hemolítica com o uso concomitante de hidroclorotiazida e metildopa.
Ciclosporina:
A terapêutica concomitante com ciclosporina pode aumentar o risco de hiperuricemia e complicações do tipo gota.
Tetraciclinas:
A administração concomitante de tetraciclinas e tiazidas aumenta o risco de uricemia induzida por tetraciclina. Provavelmente, no caso da doxiciclina esta interação não é aplicável.
Gravidez
A administração de Zolnor HCT está contraindicada durante o segundo e terceiro trimestres de gravidez (ver secções 4.3 e 4.4). Devido aos efeitos sobre a gravidez de cada uma das substâncias ativas deste medicamento, a utilização de Zolnor HCT não é recomendada durante o primeiro trimestre de gravidez (ver secção 4.4).
Olmesartan medoxomilo
A administração de ARA não é recomendada durante o primeiro trimestre de gravidez (ver secção 4.4). A administração de ARA está contraindicada durante o segundo e terceiro trimestres de gravidez (ver secções 4.3 e 4.4).
A evidência epidemiológica relativa ao risco de teratogenicidade após a exposição aos IECA durante o 1º trimestre de gravidez não é conclusiva; contudo, não é possível excluir um ligeiro aumento do risco. Enquanto não existem dados de estudos epidemiológicos controlados relativos ao risco associado aos antagonistas dos recetores da angiotensina (ARA), os riscos para esta classe de fármacos poderão ser semelhantes. A não ser que a manutenção do tratamento com ARA seja considerada essencial, nas doentes que planeiem engravidar a medicação deve ser substituída por terapêuticas anti-hipertensoras alternativas cujo perfil de segurança durante a gravidez esteja estabelecido. Quando é diagnosticada a gravidez, o tratamento com ARA deve ser interrompido imediatamente e, se apropriado, deverá ser iniciada terapêutica alternativa.
A exposição a ARA durante o segundo e terceiro trimestres de gravidez está reconhecidamente associada à indução de toxicidade fetal em humanos (diminuição da função renal, oligohidrâmnio, atraso na ossificação do crânio) e toxicidade neonatal (insuficiência renal, hipotensão, hipercaliemia) (ver secção 5.3).
No caso de a exposição a ARA ter ocorrido a partir do segundo trimestre de gravidez, recomenda-se a monitorização ultrassonográfica da função renal e dos ossos do crânio.
Recém-nascidos cujas mães estiveram expostas a ARA devem ser cuidadosamente observados no sentido de diagnosticar hipotensão (ver secções 4.3 e 4.4).
Hidroclorotiazida
A experiência decorrente da administração da hidroclorotiazida durante a gravidez, particularmente durante o primeiro trimestre, é limitada. Os estudos em animais são insuficientes.
A hidroclorotiazida atravessa a barreira placentária. Com base no mecanismo de ação farmacológico da hidroclorotiazida, a sua administração durante o segundo e o terceiro trimestres pode comprometer a perfusão fetoplacentária e pode causar efeitos fetais e neonatais tais como icterícia, distúrbios no equilíbrio eletrolítico e trombocitopenia.
A hidroclorotiazida não deve ser administrada no edema gestativo, hipertensão da gravidez ou pré-eclâmpsia devido ao risco de diminuição do volume plasmático e hipoperfusão placentária, sem efeitos benéficos relativamente ao curso da doença.
A hidroclorotiazida não deve ser administrada na hipertensão essencial em mulheres grávidas, exceto nas raras situações em que não pode ser utilizada outra alternativa terapêutica.
Amlodipina
Os dados relativos a um número limitado de gravidezes expostas não indicam que a amlodipina ou outro antagonista dos recetores de cálcio tenham um efeito nefasto na saúde do feto. Contudo, pode haver um risco de parto prolongado.
Amamentação
Uma vez que não se encontra disponível informação sobre a utilização de Zolnor HCT durantea amamentação, a terapêutica com Zolnor HCT não está recomendada e são preferíveis terapêuticas alternativas cujo perfil de segurança durante a amamentação esteja estabelecida, particularmente em recém-nascidos e pré-termo.
O olmesartan é excretado no leite de ratos lactantes. No entanto, desconhece-se se é excretado no leite humano. Desconhece-se se a amlodipina é excretada no leite materno. Semelhantes bloqueadores dos canais de cálcio do tipo di-hidropiridina são excretados no leite materno.
A hidroclorotiazida é excretada no leite humano em pequenas quantidades. As tiazidas em doses elevadas causando uma intensa diurese podem inibir a produção de leite. A utilização de Zolnor HCT durante a amamentação não é recomendada. Se Zolnor HCT for utilizado durante o aleitamento, as doses devem ser mantidas o mais baixas possível.
Fertilidade
Foram notificados casos de alterações bioquímicas reversíveis na cabeça dos espermatozoides em alguns doentes tratados com bloqueadores dos canais de cálcio. Os dados clínicos sobre o potencial efeito da amlodipina na fertilidade são insuficientes. Num estudo efetuado em ratos foram detetados efeitos adversos na fertilidade de ratos machos (ver secção 5.3).
Não foram estudados os efeitos sobre a capacidade de conduzir e utilizar máquinas. No entanto, deve ter-se em consideração que podem ocorrer ocasionalmente tonturas, cefaleias, náuseas ou fadiga em doentes submetidos a terapêutica anti- hipertensora e que estes sintomas podem diminuir a capacidade de reação. Recomenda-se precaução, especialmente no início do tratamento.
A segurança de Zolnor HCT foi estudada em ensaios clínicos em 7826 doentes a receber olmesartan medoxomilo em combinação com amlodipina e hidroclorotiazida.
As reações adversas provenientes de ensaios clínicos, estudos de segurança pós- autorização e notificação espontânea estão resumidas na tabela 1 para Zolnor HCT, bem como para os componentes individuais olmesartan medoxomilo, amlodipina e hidroclorotiazida com base no perfil de segurança conhecido de cada componente individual.
As reações adversas notificadas mais frequentemente durante o tratamento com Zolnor HCT são edema periférico, cefaleias e tonturas.
Foi utilizada a seguinte terminologia para classificar a ocorrência de efeitos indesejáveis:
Muito frequentes (≥1/10)
Frequentes (≥1/100, <1/10)
Pouco frequentes (≥1/1.000, <1/100)
Raros (≥1/10.000, <1/1.000)
Muito raros (<1/10.000)
Desconhecido (não pode ser calculado a partir dos dados disponíveis)
Tabela 1: Síntese das reações adversas com Zolnor HCT e com os componentes individuais

Foram notificados casos isolados de rabdomiólise em associação temporal com a toma de bloqueadores dos recetores da angiotensina II. Foram notificados casos isolados de síndrome extrapiramidal em doentes tratados com amlodipina.
Outras reações adversas notificadas em ensaios clínicos ou provenientes da experiência pós-comercialização com uma combinação de dose fixa de olmesartan medoxomilo e amlodipina, e ainda não notificadas para Zolnor HCT, olmesartan medoxomilo em monoterapia ou amlodipina em monoterapia ou notificadas com uma frequência mais elevada para a combinação dupla (Tabela 2):
Tabela 1: Síntese das reações adversas com Zolnor HCT e com os componentes individuais
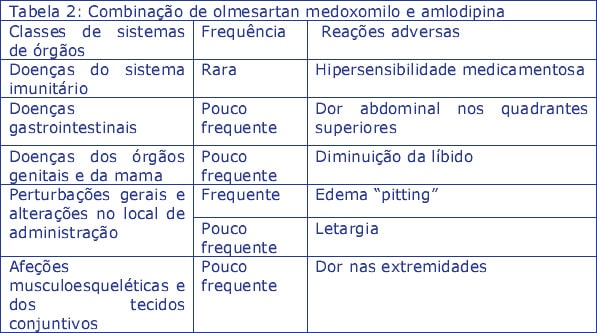
Outras reações adversas notificadas em ensaios clínicos ou provenientes da
experiência pós-comercialização com uma combinação de dose fixa de olmesartan medoxomilo e hidroclorotiazida, e ainda não notificadas para Zolnor HCT, olmesartan medoxomilo em monoterapia ou hidroclorotiazida em monoterapia ou notificadas com uma frequência mais elevada para a combinação dupla (Tabela 3):
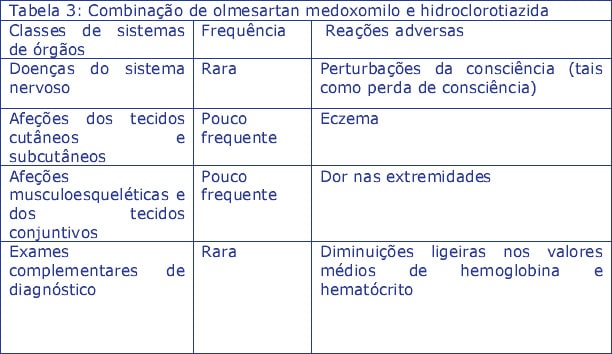
Cancro da pele não-melanoma: Com base nos dados disponíveis de estudos
epidemiológicos observou-se uma associação entre a HCTZ e o NMSC, dependente da dose cumulativa (ver também secções 4.4 e 5.1).
Notificação de suspeitas de reações adversas
A notificação de suspeitas de reações adversas após a autorização do medicamento é importante, uma vez que permite uma monitorização contínua da relação benefício- risco do medicamento. Pede-se aos profissionais de saúde que notifiquem quaisquer suspeitas de reações adversas através de:
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram
(preferencialmente)
Ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
Sintomas:
A dose máxima de Zolnor HCT é 40 mg+10 mg+25 mg uma vez por dia. Não existe informação disponível sobre a sobredosagem com Zolnor HCT em humanos. O efeito mais provável de uma sobredosagem com Zolnor HCT é hipotensão.
As manifestações mais prováveis de uma sobredosagem com olmesartan medoxomilo são hipotensão e taquicardia; podendo também verificar-se bradicardia se ocorrer estimulação parassimpática (vagal).
A sobredosagem com amlodipina pode resultar em vasodilatação periférica excessiva com hipotensão acentuada e, possivelmente, taquicardia reflexa. Foram descritas hipotensão sistémica acentuada e potencialmente prolongada até, e incluindo, choque com desfecho fatal.
Foi reportado com frequência rara edema pulmonar não cardiogénico como consequência de sobredosagem com amlodipina, que se pode manifestar com início retardado (24-48 horas após a ingestão) e requer suporte ventilatório. Medidas de reanimação precoces (incluindo sobrecarga de volume) podem ser fatores fundamentais para a manutenção da perfusão e do débito cardíaco.
A sobredosagem com hidroclorotiazida está associada à depleção eletrolítica (hipocaliemia, hipocloremia) e desidratação, decorrentes de uma diurese excessiva. Os sinais e sintomas mais frequentes de sobredosagem consistem em náuseas e sonolência. A hipocaliemia poderá induzir espasmos musculares e/ou agravamento de arritmias cardíacas associadas à administração concomitante de glicosidos digitálicos ou de alguns fármacos antiarrítmicos.
Tratamento:
Se ocorrer sobredosagem com Zolnor HCT o tratamento deve ser sintomático e de suporte. A abordagem depende do período desde a ingestão e da gravidade dos sintomas.
Se a ingestão é recente pode ser considerada uma lavagem gástrica. A administração de carvão ativado a indivíduos saudáveis imediatamente ou até duas horas após a ingestão de amlodipina demonstrou diminuir significativamente a sua absorção.
A hipotensão clinicamente significativa devido a sobredosagem de Zolnor HCT requer suporte cardiovascular ativo, incluindo a monitorização cuidadosa das funções cardíaca e respiratória, elevação das extremidades, vigilância da volémia e do débito urinário. Um vasoconstritor pode auxiliar a restabelecer o tónus vascular e a tensão arterial, desde que não haja qualquer contraindicação à sua utilização. O gluconato de cálcio intravenoso pode ser benéfico na reversão dos efeitos do bloqueio dos canais de cálcio.
Os eletrólitos séricos e os níveis de creatinina deverão ser monitorizados com frequência. Se ocorrer hipotensão, o doente deverá ser colocado em decúbito dorsal, procedendo-se à administração rápida de suplementos de sal e volume.
Dada a elevada ligação da amlodipina às proteínas, não é provável que a diálise possa ser útil. Não há informação relativamente à possibilidade de diálise do olmesartan e da hidroclorotiazida.
Não foi ainda estabelecido o grau de remoção de olmesartan e de hidroclorotiazida
por hemodiálise.
Grupo farmacoterapêutico: 3.4.2.2 Aparelho cardiovascular. Anti-hipertensores. Modificadores do eixo renina angiotensina. Antagonistas dos recetores da angiotensina.
3.4.3 Aparelho cardiovascular. Anti-hipertensores. Bloqueadores da entrada do cálcio
3.4.1.1 Aparelho cardiovascular. Anti-hipertensores. Diuréticos.Tiazidas e análogos; Código ATC: C09DX03.
O Zolnor HCT é uma associação de um antagonista dos recetores da angiotensina II, o olmesartan medoxomilo, de um bloqueador dos canais de cálcio, o besilato de amlodipina e de um diurético tiazídico, a hidroclorotiazida. A combinação destes componentes tem um efeito anti-hipertensor aditivo, reduzindo a tensão arterial em maior grau do que cada componente em separado.
O olmesartan medoxomilo é um antagonista seletivo dos recetores da angiotensina II (tipo AT1), ativo por via oral. A angiotensina II é a principal hormona vasoativa do sistema renina-angiotensina-aldosterona e desempenha um papel significativo na fisiopatologia da hipertensão. Os efeitos da angiotensina II incluem vasoconstrição, estimulação da síntese e libertação de aldosterona, estimulação cardíaca e reabsorção renal de sódio. O olmesartan inibe os efeitos vasoconstrictor e secretor de aldosterona da angiotensina II por bloqueio da sua ligação ao recetor AT1 em tecidos, incluindo o músculo liso vascular e a glândula suprarrenal. A ação do olmesartan é independente da origem ou via de síntese da angiotensina II. O antagonismo seletivo em relação aos recetores da angiotensina II (tipo AT1) do olmesartan induz um aumento dos níveis plasmáticos de renina e das concentrações da angiotensina I e angiotensina II e alguma diminuição das concentrações plasmáticas da aldosterona.
Na hipertensão, o olmesartan medoxomilo induz uma diminuição da tensão arterial de longa duração e dose-dependente. Não se registou qualquer ocorrência de hipotensão após a primeira dose, de taquifilaxia durante o tratamento a longo prazo ou de hipertensão reacional após suspensão abrupta do tratamento.
A administração uma vez por dia de olmesartan medoxomilo induz uma redução eficaz e suave da tensão arterial durante um período de 24 horas. A administração uma vez por dia induziu uma redução da tensão arterial semelhante à que se verificou com a administração duas vezes por dia da mesma dose total diária.
Em tratamento continuado, as reduções máximas da tensão arterial são atingidas 8 semanas após o início da terapêutica, embora uma proporção substancial da redução da tensão arterial seja observada logo após 2 semanas de tratamento.
O efeito do olmesartan medoxomilo na mortalidade e morbilidade não é ainda conhecido.
O estudo “Randomised Olmesartan and Diabetes Microalbuminuria Prevention"
(ROADMAP), realizado em 4447 doentes com diabetes tipo 2, normoalbuminúria e pelo menos um fator de risco cardiovascular adicional, investigou se o tratamento com olmesartan podia adiar o início de microalbuminúria. Durante o período de seguimento mediano, com duração de 3,2 anos, os doentes receberam ou olmesartan ou placebo em adição a outros agentes anti-hipertensores, exceto IECAs ou ARAs.
Para o endpoint primário, o estudo demonstrou uma redução significativa do risco no tempo para início de microalbuminúria, a favor de olmesartan. Após ajuste para diferenças de pressão arterial, esta redução do risco já não era estatisticamente significativa. 8,2% (178 de 2160) dos doentes no grupo olmesartan e 9,8% (210 de 2139) dos doentes no grupo placebo desenvolveram microalbuminúria.
Em relação aos endpoints secundários, ocorreram acontecimentos cardiovasculares em 96 doentes (4,3%) com olmesartan e em 94 doentes (4,2%) com placebo. A incidência de mortalidade cardiovascular foi mais elevada com olmesartan comparativamente com o tratamento placebo (15 doentes (0,7%) vs. 3 doentes (0,1%)), apesar de as taxas para acidente vascular cerebral não fatal (14 doentes (0,6%) vs. 8 doentes (0,4%)), enfarte do miocárdio não fatal (17 doentes (0,8%) vs. 26 doentes (1,2%)) e mortalidade não cardiovascular (11 doentes (0,5%) vs. 12 doentes (0,5%)) serem similares. A mortalidade global com olmesartan aumentou numericamente (26 doentes (1,2%) vs. 15 doentes (0,7%)), o que foi principalmente impulsionado por um número mais elevado de acontecimentos cardiovasculares fatais.
O estudo “Olmesartan Reducing Incidence of End-stage Renal Disease in Diabetic Nephropathy Trial" (ORIENT) investigou os efeitos do olmesartan nos resultados renais e cardiovasculares em 577 doentes japoneses e chineses, aleatorizados, com diabetes tipo 2 e com nefropatia evidente. Durante um período de seguimento mediano de 3,1 anos, os doentes receberam ou olmesartan ou placebo em adição a outros agentes anti-hipertensores, incluindo IECAs.
O endpoint primário composto (tempo até ao primeiro acontecimento de duplicação da creatinina sérica, doença renal terminal, morte por todas as causas) ocorreu em 116 doentes no grupo olmesartan (41,1%) e 129 doentes no grupo placebo (45,4%) (HR 0,97 (95% IC 0,75 a 1,24); p=0,791). O endpoint cardiovascular secundário composto ocorreu em 40 doentes tratados com olmesartan (14,2%) e 53 doentes tratados com placebo (18,7%). Este endpoint cardiovascular composto incluiu morte cardiovascular em 10 (3,5%) doentes a tomar olmesartan versus 3 (1,1%) doentes a tomar placebo, mortalidade global 19 (6,7%) versus 20 (7,0%), acidente vascular cerebral não fatal 8 (2,8%) versus 11 (3,9%) e enfarte do miocárdio não fatal 3 (1,1%) versus 7 (2,5%), respetivamente.
O componente amlodipina do Zolnor HCT é um bloqueador dos canais de cálcio que inibe a entrada transmembranária de iões de cálcio, através dos canais tipo L dependentes da voltagem, no coração e no músculo liso. Dados experimentais sugerem que a amlodipina se liga a ambos os locais de ligação, dihidropiridina e não- di-hidropiridina. A amlodipina é relativamente vaso-seletiva, com um maior efeito nas células do músculo liso vascular do que nas células do músculo cardíaco. O efeito anti-hipertensor da amlodipina deve-se a um efeito relaxante direto no músculo liso arterial, que provoca reduções na resistência periférica e na tensão arterial.
Em doentes hipertensos, a amlodipina induz uma diminuição da tensão arterial de longa duração e dose-dependente. Não se registou qualquer ocorrência de hipotensão após a primeira dose, de taquifilaxia durante o tratamento a longo prazo ou de hipertensão reativa após suspensão abrupta do tratamento.
Após administração de doses terapêuticas a doentes hipertensos, a amlodipina produz uma redução efetiva da tensão arterial nas posições supina, sentada e ortostática. O uso crónico de amlodipina não está associado a alterações significativas da frequência cardíaca ou dos níveis plasmáticos de catecolaminas. Em doentes hipertensos com função renal normal, doses terapêuticas de amlodipina reduzem a resistência vascular renal e aumentam a taxa de filtração glomerular e o fluxo plasmático renal efetivo, sem alteração na fração de filtração ou na proteinúria.
Estudos hemodinâmicos em doentes com insuficiência cardíaca e estudos clínicos baseados em testes de exercício em doentes com insuficiência cardíaca de classe II- IV da NYHA, mostraram que a amlodipina não provoca qualquer descompensação clínica, avaliada pela tolerância ao exercício, fração de ejeção ventricular esquerda e sinais e sintomas clínicos.
Um estudo controlado com placebo (PRAISE), desenhado para avaliar doentes com insuficiência cardíaca de classe III-IV da NYHA a receber digitálicos, diuréticos e IECAs, mostrou que a amlodipina não levou a um aumento no risco de mortalidade e de morbilidade em doentes com insuficiência cardíaca.
Num estudo de seguimento de longa duração, controlado com placebo (PRAISE-2) com amlodipina em doentes com insuficiência cardíaca de classe III e IV da NYHA, sem sintomas clínicos ou dados objetivos sugestivos de doença isquémica subjacente, com doses estáveis de IECAs, digitálicos e diuréticos, a amlodipina não demonstrou ter efeito na mortalidade total ou cardiovascular. Nesta mesma população, a amlodipina foi associada a um número crescente de notificações de edema pulmonar apesar de não ter sido verificada uma diferença significativa na incidência de agravamento da insuficiência cardíaca quando comparada com o placebo.
Foi realizado um estudo de morbilidade e mortalidade, aleatorizado, em dupla ocultação denominado “The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Tria" (ALLHAT), com o objetivo de comparar terapêuticas mais recentes: amlodipina 2,5-10 mg/dia (bloqueador dos canais de cálcio) ou lisinopril 10-40 mg/dia (inibidor da ECA) como tratamentos de primeira linha, relativamente a uma terapêutica com umdiurético tiazídico, a clorotalidona 12,5-25 mg/dia, na hipertensão ligeira a moderada.
Foram aleatorizados um total de 33.357 doentes hipertensos com idade igual ou superior a 55 anos que foram seguidos durante uma média de 4,9 anos. Os doentes tinham pelo menos um fator de risco adicional para a doença coronária, incluindo: enfarte do miocárdio ou acidente vascular cerebral prévios (> 6 meses antes do recrutamento) ou outra doença cardiovascular aterosclerótica documentada (no total 51,5%), diabetes tipo 2 (36,1%), colesterol-HDL < 35 mg/dl (11,6%), hipertrofia ventricular esquerda diagnosticada por eletrocardiograma ou ecocardiografia (20,9%), hábitos tabágicos atuais (21,9%).
O parâmetro de avaliação primário era composto por doença coronária fatal ou enfarte do miocárdio não fatal. Não houve diferença significativa no parâmetro de avaliação primário entre a terapêutica com amlodipina e a terapêutica com clorotalidona: RR 0,98 95% IC (0,90-1,07) p=0,65. Entre os parâmetros de avaliação secundários, a incidência de insuficiência cardíaca (componente do desfecho de um parâmetro de avaliação cardiovascular combinado composto) foi significativamente superior no grupo da amlodipina quando comparado com o grupo da clorotalidona (10,2% vs. 7,7%, RR 1,38, 95% IC [1,25-1,52] p<0,001). No entanto, não houve diferença significativa na mortalidade por todas as causas entre a terapêutica com amlodipina e a terapêutica com clorotalidona. RR 0,96 95% IC [0,89-1,02] p=0,20.
A hidroclorotiazida é um diurético tiazídico. O mecanismo do efeito anti-hipertensor dos diuréticos tiazídicos não é totalmente conhecido. As tiazidas interferem com o mecanismo tubular renal de reabsorção dos eletrólitos, aumentando diretamente a excreção de sódio e de cloreto em quantidades aproximadamente equivalentes. A ação diurética da hidroclorotiazida reduz o volume plasmático, aumenta a atividade plasmática da renina e aumenta a secreção de aldosterona, com aumento consequente de potássio urinário e perda de bicarbonato e diminuição de potássio sérico. A relação renina-aldosterona é mediada pela angiotensina II e consequentemente a administração concomitante de um antagonista dos recetores da angiotensina II tende a reverter a perda de potássio associada aos diuréticos tiazídicos. Com a hidroclorotiazida, o início da diurese ocorre cerca de 2 horas após a administração e o efeito máximo ocorre cerca de 4 horas após a administração, enquanto que a ação persiste durante cerca de 6 a 12 horas.
Estudos epidemiológicos demonstraram que o tratamento a longo prazo com hidroclorotiazida em monoterapia reduz o risco de mortalidade e morbilidade cardiovascular.
Resultados de Estudos Clínicos
Num estudo em 2492 doentes (67% Caucasianos), com a duração de 12 semanas, com grupos paralelos, aleatorizado e sob dupla ocultação, o tratamento com Zolnor HCT 40 mg+10 mg+ 25 mg resultou em significativamente maiores reduções da tensão arterial diastólica e sistólica do que o tratamento com cada uma das combinações duplas correspondentes, olmesartan medoxomilo 40 mg mais amlodipina 10 mg, olmesartan medoxomilo 40 mg mais hidroclorotiazida 25 mg e amlodipina 10 mg mais hidroclorotiazida 25 mg, respetivamente.
O efeito adicional de redução da tensão arterial de Zolnor HCT 40 mg+10 mg+25 mg quando comparado com as combinações duplas análogas foi de entre -3,8 e -6,7 mmHg para a tensão arterial diastólica em posição sentada e de entre -7,1 e -9,6 mmHg para a tensão arterial sistólica em posição sentada, tendo este efeito ocorrido nas duas primeiras semanas de tratamento.
As proporções de doentes que alcançaram a tensão arterial alvo (<140/90 mmHg para doentes não diabéticos e <130/80 mmHg para doentes diabéticos) às 12 semanas variaram de 34,9% a 46,6% para os grupos submetidos ao tratamento com a combinação dupla, comparativamente com 64,3% para Zolnor HCT 40 mg+10 mg+25 mg.
Num segundo estudo em 2690 doentes (99,9% caucasianos) com grupos paralelos, aleatorizado e sob dupla ocultação, o tratamento com Zolnor HCT (20 mg + 5 mg + 12,5, 40 mg + 5 mg + 12,5 mg, 40 mg + 5 mg + 25 mg, 40 mg + 10 mg + 12,5 mg, 40 mg + 10 mg + 25 mg) resultou em significativamente maiores reduções da tensão arterial diastólica e sistólica, quando comparado com as combinações duplas correspondentes, olmesartan medoxomilo 20 mg mais amlodipina 5 mg, olmesartan medoxomilo 40 mg mais amlodipina 5 mg e olmesartan medoxomilo 40 mg mais amlodipina 10 mg, respetivamente, após 10 semanas de tratamento.
O efeito adicional de redução da tensão arterial de Zolnor HCT quando comparado com as combinações duplas correspondentes foi de entre -1,3 e -1,9 mmHg para a tensão arterial diastólica em posição sentada e de entre - 2,7 e - 4,9 mmHg para a tensão arterial sistólica em posição sentada.
As proporções de doentes que alcançaram a tensão arterial alvo (<140/90 mmHg para doentes não diabéticos e <130/80 mmHg para doentes diabéticos) às 10 semanas variaram de 42,7% a 49,6% para os grupos submetidos ao tratamento com a combinação dupla, comparativamente com 52,4% a 58,8% para Zolnor HCT.
Num estudo add-on aleatorizado, sob dupla ocultação em 808 doentes (99,9% caucasianos) não adequadamente controlados após 8 semanas de terapêutica com olmesartan medoxomilo 40 mg mais amlodipina 10 mg em combinação dupla, o tratamento com Zolnor HCT resultou numa redução numericamente adicional da tensão arterial em posição sentada de -1,8 / -1,0 mmHg quando tratados com Zolnor HCT 40 mg + 10 mg + 12,5 mg e numa redução adicional estatisticamente significativa da tensão arterial em posição sentada de -3,6 / -2,8 mmHg quando tratados com Zolnor HCT 40 mg + 10 mg + 25 mg, comparativamente com olmesartan medoxomilo 40 mg mais amlodipina 10 mg em combinação dupla.
O tratamento com a combinação tripla Zolnor HCT 40 mg + 10 mg + 25 mg resultou numa percentagem superior estatisticamente significativa de indivíduos que atingem o seu objetivo de tensão arterial comparativamente com olmesartan medoxomilo 40 mg mais amlodipina 10 mg em combinação dupla (41,3% vs. 24,2%); enquanto que o tratamento com a combinação tripla Zolnor HCT 40 mg + 10 mg + 12,5 mg resultou numa percentagem numericamente superior de indivíduos que atingem o seu objetivo de tensão arterial comparativamente com olmesartan medoxomilo 40 mg mais amlodipina 10 mg em combinação dupla (29,5% vs. 24,2%) em indivíduos não adequadamente controlados com a terapêutica com a combinação dupla.
O efeito anti-hipertensor de Zolnor HCT foi similar independentemente da idade e género e foi similar em doentes com ou sem diabetes.
Outra informação:
Dois grandes estudos aleatorizados e controlados (ONTARGET (“ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial") e VA NEPHRON-D (“The Veterans Affairs Nephropathy in Diabetes")) têm examinado o uso da associação de um inibidor da ECA com um antagonista dos recetores da angiotensina II.
O estudo ONTARGET foi realizado em doentes com história de doença cardiovascular ou cerebrovascular, ou diabetes mellitus tipo 2 acompanhada de evidência de lesão de órgão-alvo. O estudo VA NEPHRON-D foi conduzido em doentes com diabetes mellitus tipo 2 e nefropatia diabética.
Estes estudos não mostraram nenhum efeito benéfico significativo nos resultados renais e/ou cardiovasculares e mortalidade, enquanto foi observado um risco aumentado de hipercaliemia, insuficiência renal aguda e/ou hipotensão, em comparação com monoterapia. Dadas as suas propriedades farmacodinâmicas semelhantes, estes resultados são também relevantes para outros inibidores da ECA e antagonistas dos recetores da angiotensina II.
Os inibidores da ECA e os antagonistas dos recetores da angiotensina II não devem assim, ser utilizados concomitantemente em doentes com nefropatia diabética.
O estudo ALTITUDE (“Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints") foi concebido para testar o benefício da adição de aliscireno a uma terapêutica padrão com um inibidor da ECA ou um antagonista dos recetores da angiotensina II em doentes com diabetes mellitus tipo 2 e doença renal crónica, doença cardiovascular ou ambas. O estudo terminou precocemente devido a um risco aumentado de resultados adversos. A morte cardiovascular e o acidente vascular cerebral foram ambos numericamente mais frequentes no grupo tratado com aliscireno, do que no grupo tratado com placebo e os acontecimentos adversos e acontecimentos adversos graves de interesse (hipercaliemia, hipotensão e disfunção renal) foram mais frequentemente notificados no grupo tratado com aliscireno que no grupo tratado com placebo.
Cancro da pele não-melanoma:
Com base nos dados disponíveis de estudos epidemiológicos, observou-se uma associação entre a HCTZ e o NMSC, dependente da dose cumulativa. Um estudo incluiu uma população constituída por 71 533 casos de BCC e por 8 629 casos de SCC, em 1 430 833 e 172 462 controlos, respetivamente, da população em estudo. Uma utilização elevada de HCTZ (≥50 000 mg cumulativos) foi associada a uma taxa de probabilidade (OR) ajustada de 1,29 (95 % IC: 1,23-1,35) para BCC e 3,98 (95 % IC: 3,68-4,31) para SCC. Observou-se uma clara relação da resposta à dose cumulativa para BCC e SCC. Outro estudo revelou uma possível associação entre o carcinoma espinocelular (SCC) do lábio e a exposição à HCTZ: 633 casos de SCC do lábio foram identificados em 63 067 controlos da população, com base numa amostragem longitudinal (risk-set sampling). Foi demonstrada uma associação dose- resposta com uma taxa de probabilidade (OR) ajustada de 2,1 (95 % IC: 1,7-2,6), aumentando OR para 3,9 (95 % IC: 3,0-4,9) para uma utilização elevada (25 000 mg HCTZ) e para OR de 7,7 (95 % IC: 5,7-10,5) para a dose cumulativa mais elevada (aprox.100 000 mg HCTZ) (ver também secção 4.4).
A administração concomitante de olmesartan medoxomilo, amlodipina e hidroclorotiazida não teve efeitos clinicamente relevantes na farmacocinética de cada componente em indivíduos saudáveis.
Após a administração oral de Zolnor HCT em adultos saudáveis normais, o pico da concentração plasmática de olmesartan, amlodipina e hidroclorotiazida é alcançado em cerca de 1,5 a 3 h, 6 a 8 h e 1,5 a 2 horas, respetivamente. A taxa e a extensão da absorção de olmesartan medoxomilo, amlodipina e hidroclorotiazida do Zolnor HCT são iguais às obtidas aquando da administração sob a forma de uma combinação dupla fixa de olmesartan medoxomilo e amlodipina juntamente com um comprimido com o componente hidroclorotiazida, ou aquando da administração sob a forma de uma combinação dupla fixa de olmesartan medoxomilo e hidroclorotiazida juntamente com um comprimido com o componente amlodipina nas mesmas dosagens. Os alimentos não afetam a biodisponibilidade do Zolnor HCT.
Olmesartan medoxomilo:
Absorção e distribuição:
O olmesartan medoxomilo é um pró-fármaco. É rapidamente convertido no metabolito farmacologicamente ativo, olmesartan, por esterases na mucosa intestinal e no sangue portal, durante a absorção pelo trato gastrointestinal. O olmesartan medoxomilo ou a fração molecular medoxomilo da cadeia lateral não foram detetados intactos no plasma ou excreções. A biodisponibilidade absoluta média do olmesartan na formulação de comprimidos foi de 25,6%.
O pico médio de concentração plasmática (Cmax) de olmesartan é atingido cerca de 2 horas após a administração oral de olmesartan medoxomilo e as concentrações plasmáticas de olmesartan aumentam de forma quase linear com doses orais únicas crescentes até cerca de 80 mg.
Os alimentos tiveram um efeito mínimo na biodisponibilidade do olmesartan, pelo que o olmesartan medoxomilo pode ser administrado com ou sem alimentos.
Não foram observadas diferenças clinicamente relevantes na farmacocinética do olmesartan relacionadas com o sexo.
O olmesartan apresenta uma forte ligação às proteínas plasmáticas (99,7%), no entanto, o potencial para originar interações de deslocação clinicamente significativas entre o olmesartan e outros fármacos administrados concomitantemente com uma forte ligação às proteínas plasmáticas é baixo (o que se comprova pela ausência de interações clinicamente significativas entre o olmesartan medoxomilo e a varfarina). A ligação do olmesartan às células sanguíneas é insignificante. O volume médio de distribuição após a administração intravenosa é baixo (16 – 29 l).
Biotransformação e eliminação:
A depuração plasmática total foi de 1,3 l/h (coeficiente de variação, 19%) e foi relativamente lenta quando comparada com o fluxo sanguíneo hepático (cerca de 90 l/h). Após uma dose oral única de olmesartan medoxomilo marcado com 14C, 10 a 16% da radioatividade administrada foi excretada na urina (a grande maioria nas 24 horas após a administração da dose) e a restante radioatividade recuperada foi excretada nas fezes. Considerando a disponibilidade sistémica de 25,6%, pode calcular-se que o olmesartan absorvido é eliminado por excreção renal (cerca de 40%) e hepatobiliar (cerca de 60%). A radioatividade recuperada foi totalmente identificada como olmesartan. Não foi detetado qualquer outro metabolito significativo. A recirculação entero-hepática do olmesartan é mínima. Uma vez que uma grande proporção de olmesartan é excretada por via biliar, a utilização em doentes com obstrução biliar é contraindicada (ver secção 4.3).
A semivida de eliminação terminal do olmesartan variou entre 10 e 15 horas após administração de doses orais múltiplas. O estado estacionário foi atingido após 2-5 dias de administração e não se observou acumulação adicional 14 dias após a administração repetida. A depuração renal foi aproximadamente de 0,5 a 0,7 l/h e foi independente da dose.
Interações medicamentosas
Agente sequestrador de ácidos biliares, colessevelam:
A administração concomitante de 40 mg de olmesartan medoxomilo e de 3750 mg de cloridrato de colessevelam em indivíduos saudáveis, resultou numa redução de 28% na Cmax e numa redução de 39% na AUC de olmesartan. Foram observados efeitos menores, reduções de 4% e de 15% na Cmax e AUC, respetivamente, quando o olmesartan medoxomilo foi administrado 4 horas antes do cloridrato de colessevelam. A semivida de eliminação de olmesartan foi reduzida em 50 - 52% independentemente de ter sido administrado concomitantemente, ou 4 horas antes do cloridrato de colessevelam (ver secção 4.5).
Amlodipina:
Absorção e distribuição:
Após administração oral de doses terapêuticas, a amlodipina é bem absorvida com picos séricos entre 6 a 12 horas após a dose. Estima-se que a biodisponibilidade absoluta seja entre 64 e 80%. O volume de distribuição é de aproximadamente 21 l/kg. Estudos in vitro mostraram que aproximadamente 97,5% da amlodipina em circulação está ligada a proteínas plasmáticas.
A absorção da amlodipina não é afetada pela ingestão concomitante de alimentos.
Biotransformação e eliminação:
A semivida de eliminação plasmática terminal é de 35 a 50 horas e é consistente com uma única toma diária.
A amlodipina é extensivamente metabolizada pelo fígado em metabolitos inativos sendo de 10% a eliminação urinária da amlodipina e de 60% a eliminação urinária dos metabolitos.
Hidroclorotiazida:
Absorção e distribuição:
Após a administração oral de olmesartan medoxomilo e hidroclorotiazida em combinação, o tempo médio para atingir o pico das concentrações de hidroclorotiazida foi de 1,5 a 2 horas após a administração. A hidroclorotiazida tem uma ligação às proteínas plasmáticas de 68% e o seu volume de distribuição aparente é de 0,83 – 1,14 l/kg.
Biotransformação e eliminação:
A hidroclorotiazida não é metabolizada no homem e é excretada quase completamente como fármaco inalterado na urina. Cerca de 60% da dose oral é eliminada como fármaco inalterado em 48 horas. A depuração renal é cerca de 250 – 300 ml/min. A semivida de eliminação terminal da hidroclorotiazida é de 10 a 15 horas.
Farmacocinética em populações especiais
População pediátrica:
A Agência Europeia do Medicamento dispensou a obrigação de submissão de resultados de estudos com Zolnor HCT para o tratamento da hipertensão essencial em todos os subgrupos de população pediátrica.
Idosos (idade igual ou superior a 65 anos):
Em doentes hipertensos, a AUC do olmesartan no estado estacionário aumentou em cerca de 35% em doentes idosos (65 – 75 anos) e em cerca de 44% em muito idosos (> 75 anos) em comparação com o grupo etário mais jovem (ver secção 4.2). Isto pode estar, pelo menos em parte, relacionado com uma redução média da função renal neste grupo de doentes. O regime posológico recomendado para idosos é, porém, o mesmo, embora se deva ter precaução quando se aumenta a dose.
O tempo para atingir o pico de concentrações plasmáticas de amlodipina é idêntico em indivíduos idosos e jovens. Em doentes idosos, a depuração da amlodipina tende a diminuir, resultando em aumentos da AUC e da semivida de eliminação. Os aumentos da AUC e da semivida de eliminação em doentes com insuficiência cardíaca congestiva foram os esperados para o grupo etário dos doentes neste estudo (ver secção 4.4).
Dados ainda limitados sugerem que a depuração sistémica da hidroclorotiazida é inferior em doentes idosos, quer saudáveis quer hipertensos, em comparação com voluntários jovens e saudáveis.
Compromisso renal:
Em doentes com compromisso renal, a AUC de olmesartan no estado estacionário aumentou 62%, 82% e 179% em doentes com compromisso renal ligeiro, moderado e grave respetivamente, em comparação com os controlos saudáveis (ver secções 4.2 e 4.4). A farmacocinética do olmesartan medoxomilo em doentes submetidos a hemodiálise não foi estudada.
A amlodipina é extensivamente metabolizada em metabolitos inativos. 10% da substância é excretada inalterada na urina. As alterações nas concentrações plasmáticas da amlodipina não estão correlacionadas com o grau de compromisso renal. Nestes doentes, a amlodipina pode ser administrada em doses normais. A amlodipina não é dialisável.
A semivida da hidroclorotiazida é prolongada em doentes com compromisso renal.
Compromisso hepático:
Após administração oral única, os valores da AUC de olmesartan foram 6% e 65% mais elevados em doentes com compromisso hepático ligeiro e moderado, respetivamente, do que nos correspondentes controlos saudáveis. A fração livre de olmesartan 2 horas após a administração a indivíduos saudáveis, doentes com compromisso hepático ligeiro e doentes com compromisso hepático moderado foi, respetivamente, 0,26%, 0,34% e 0,41%.
Após doses repetidas em doentes com disfunção hepática moderada, os valores médios da AUC de olmesartan foram, novamente, cerca de 65% mais elevados do que nos seus correspondentes controlos saudáveis. Os valores médios da Cmax de olmesartan foram similares nos doentes com compromisso hepático e nos indivíduos saudáveis.
O olmesartan medoxomilo não foi avaliado em doentes com compromisso hepático grave (ver secções 4.2 e 4.4).
Os doentes com disfunção hepática apresentam uma reduzida depuração da amlodipina e um prolongamento da semivida, resultando num aumento da AUC em cerca de 40% - 60% (ver secções 4.2 e 4.4).
A informação clínica disponível sobre a administração da amlodipina em doentes com compromisso hepático é muito limitada.
O compromisso hepático não influencia significativamente a farmacocinética da hidroclorotiazida.
Associação Olmesartan medoxomilo + Amlodipina + Hidroclorotiazida
Num estudo de toxicidade de dose repetida, em ratos, foi demonstrado que a administração em associação de olmesartan medoxomilo, amlodipina e hidroclorotiazida não aumentou nenhuma das toxicidades previamente existentes e reportadas dos agentes individuais, nem induziu nenhuma nova toxicidade. Também não foram observados efeitos toxicológicos sinérgicos.
Não foram realizados estudos adicionais de mutagenicidade, carcinogenicidade e toxicidade reprodutiva para o Zolnor HCT.
Olmesartan medoxomilo
Em estudos de toxicidade crónica em ratos e cães, o olmesartan medoxomilo evidenciou efeitos semelhantes aos dos outros antagonistas dos recetores AT1 e inibidores da ECA: ureia sanguínea (BUN) e creatinina elevadas, diminuição do peso cardíaco, diminuição dos parâmetros eritrocitários (eritrócitos, hemoglobina, hematócrito), indicações histológicas de lesão renal (lesões regenerativas do epitélio renal, espessamento da membrana basal, dilatação dos túbulos). Estes efeitos adversos causados pela ação farmacológica do olmesartan medoxomilo também ocorreram em ensaios pré-clínicos com outros antagonistas dos recetores AT1 e com outros inibidores da ECA e podem ser diminuídos pela administração oral simultânea de cloreto de sódio.
Tal como outros antagonistas dos recetores AT1, o olmesartan medoxomilo aumentou a incidência de ruturas cromossómicas em culturas celulares in vitro, mas não in vivo. Os dados globais de uma série extensiva de testes de genotoxicidade sugerem que é muito improvável que o olmesartan exerça efeitos genotóxicos nas condições de uso clínico.
O olmesartan medoxomilo não se revelou carcinogénico em ratos ou ratinhos usando modelos transgénicos.
Em estudos de reprodução em ratos, o olmesartan medoxomilo não afetou a fertilidade e não houve qualquer evidência de efeito teratogénico. Em comum com outros antagonistas da angiotensina II, a sobrevida da descendência diminuiu e observou-se dilatação pélvica renal após exposição das progenitoras na fase tardia da gestação e lactação. Em coelhos não houve qualquer indicação de efeito fetotóxico.
Amlodipina
Toxicologia reprodutiva
Estudos de reprodução em ratos e ratinhos mostraram um atraso na data de parto, da duração prolongada do trabalho de parto e diminuição da sobrevivência das crias, em doses aproximadamente 50 vezes superiores à dose máxima recomendada para humanos, com base em mg/kg.
Compromisso da fertilidade
Não houve efeito na fertilidade de ratos tratados com amlodipina (machos durante 64 dias e fêmeas 14 dias antes do acasalamento) em doses até 10 mg/kg/dia (8 vezes* a dose máxima recomendada de 10 mg para o humano com base em mg/m2). Noutro estudo com ratos, no qual os ratos machos foram tratados com besilato de amlodipina durante 30 dias com uma dose comparável à dose humana com base em mg/kg, foi observada uma diminuição plasmática da hormona folículo- estimulante e da testosterona, assim como uma diminuição da densidade do esperma e do número de espermatídeos maduros e células de Sertoli.
Carcinogénese, mutagénese
Ratos e ratinhos tratados com amlodipina na dieta durante dois anos, em concentrações calculadas para proporcionarem níveis de dose diária de 0,5, 1,25 e 2,5 mg/kg/dia não mostraram evidência de carcinogenicidade. A dose mais elevada (para ratinhos, semelhante à dose máxima recomendada de 10 mg com base em mg/m2 e para ratos o dobro dessa dose*) foi próxima da dose máxima tolerada para os ratinhos, mas não para os ratos.
Estudos de mutagénese não demonstraram efeitos relacionados com o medicamento tanto ao nível dos genes como dos cromossomas.
*Baseado num peso de doente de 50 kg
Hidroclorotiazida
Estudos com hidroclorotiazida demonstraram evidência equívoca de um efeito genotóxico e carcinogénico em alguns modelos experimentais.
Núcleo do comprimido
Amido de milho pré-gelificado
Celulose microcristalina
Sílica Coloidal anidra
Croscarmelose sódica
Estearato de magnésio
Revestimento do comprimido
Álcool polivinílico
Macrogol 3350
Talco
Dióxido de titânio (E 171)
Óxido de ferro (III) amarelo (E 172)
Óxido de ferro (III) vermelho (E 172) (apenas os comprimidos revestidos por película 20+5+12,5; 40+10+12,5; 40+10+25)
Óxido de ferro (II, III) preto (E 172) (apenas os comprimidos revestidos por película 20+5+12,5)
Não aplicável.
3 anos.
O medicamento não necessita de quaisquer precauções especiais de conservação.
Blister laminado de poliamida/alumínio/cloreto de polivinilo/alumínio.
Tamanhos de embalagens: 14, 28, 30, 56, 84, 90, 98, 10 x 28 e 10 x 30 comprimidos revestidos por película em blisters.
10 x 1, 50 x 1 e 500 x 1 comprimidos revestidos por película em blisters destacáveis para dose unitária.
Frascos de HDPE de 30 ml com fecho de polipropileno resistente à abertura por crianças, revestido com selo interior e com sílica gel exsicante.
Embalagens de 7 e 30 comprimidos revestidos por película.
Frascos de HDPE de 60 ml com fecho de polipropileno resistente à abertura por crianças, revestido com selo interior e com sílica gel exsicante.
Embalagens de 90 comprimidos revestidos por película.
É possível que não sejam comercializadas todas as apresentações.
Não existem requisitos especiais.
Menarini International Operations Luxembourg S.A.
1, Avenue de la Gare
L-1611 Luxemburgo
Sob licença da Daiichi Sankyo Europe GmbH
N.º de registo: 5369319 – 14 comprimidos, 20 mg+5 mg+12,5 mg, blister
N.º de registo: 5369327 – 56 comprimidos, 20 mg+5 mg+12,5 mg, blister
N.º de registo: 5369335 – 14 comprimidos, 40 mg+5 mg+12,5 mg, blister
N.º de registo: 5369343 – 56 comprimidos, 40 mg+5 mg+12,5 mg, blister
N.º de registo: 5369368 – 14 comprimidos, 40 mg+5 mg+12,5 mg, blister
N.º de registo: 5369350 – 56 comprimidos, 40 mg+5 mg+12,5 mg, blister
N.º de registo: 5369376 – 14 comprimidos, 40 mg+5 mg+25 mg, blister
N.º de registo: 5369400 – 56 comprimidos, 40 mg+5 mg+25 mg, blister
N.º de registo: 5369418 – 14 comprimidos, 40 mg+10 mg+25 mg, blister
N.º de registo: 5369426 – 56 comprimidos, 40 mg+10 mg+25 mg, blister
Data da primeira autorização: 10 de março de 2011
