












ZANIPRESS 20/20 mg



Zanipress 20 mg + 20 mg comprimidos revestidos por película
Cada comprimido revestido por película contém 20 mg de maleato de enalapril (equivalente a 15,29 mg de enalapril) e 20 mg de cloridrato de lercanidipina (equivalente a 18,88 mg de lercanidipina).
Excipiente com efeito conhecido: cada comprimido contém 204 mg de lactose mono- hidratada.
Lista completa de excipientes, ver secção 6.1.
Comprimido revestido por película.
Comprimido de 12 mm, cor de laranja, circular, biconvexo.
Tratamento da hipertensão arterial, como terapêutica de substituição em doentes adultos com pressão arterial adequadamente controlada com 20 mg de enalapril e 20mg de lercanidipina, administrados isoladamente.
Posologia:
A dose recomendada é de um comprimido uma vez ao dia, pelo menos 15 minutos antes da refeição.
Idosos:
A dose a administrar deve depender da função renal do doente (ver “Utilização na insuficiência renal”).
Compromisso renal:
Zanipress está contraindicado em doentes com insuficiência renal grave (clearance da creatinina <30 ml/min) ou em doentes a fazer hemodiálise (ver secção 4.3 e 4.4). É necessária precaução particular no início do tratamento de doentes com insuficiência renal ligeira a moderada.
Compromisso hepático:
Zanipress está contraindicado na insuficiência hepática grave. É necessária precaução particular no início do tratamento de doentes com insuficiência hepática ligeira a moderada.
População pediátrica:
Não existe utilização relevante de Zanipress na população pediátrica na indicação de hipertensão.
Modo de administração:
Precauções a ter em conta antes de manusear ou administrar o medicamento:
• O tratamento deve ser administrado de preferência de manhã, pelo menos 15 minutos antes do pequeno-almoço.
• Este medicamento não deve ser administrado com sumo de toranja (ver secção 4.3 e 4.5).
• Hipersensibilidade a qualquer inibidor da enzima de conversão da angiotensina (ECA), ou a qualquer dihidropiridina bloqueadora de cálcio, ou a qualquer um dos excipientes mencionados na secção 6.1.
• História de angioedema associado a terapêutica com inibidores da enzima de conversão da angiotensina (ECA).
• Angioedema hereditário ou idiopático.
• Segundo e terceiro trimestres da gravidez (ver secção 4.4 e 4.6).
• Obstrução da via de saída do ventrículo esquerdo.
• Insuficiência cardíaca congestiva não tratada.
• Angina de peito instável ou enfarte do miocárdio recente (no período de 1 mês).
• Compromisso hepático grave.
• Compromisso renal grave (TFG <30 ml/min.), incluindo doentes a fazer diálise.
• Coadministração com:
◦ Inibidores fortes do CYP3A4 (ver secção 4.5)
◦ Ciclosporina (ver secção 4.5)
◦ Toranja ou sumo de toranja (ver secção 4.5).
• Uso concomitante com terapêutica com sacubitril / valsartan. O enalapril não deve ser iniciado antes de 36 horas após a última dose de sacubitril / valsartan (ver também secções 4.4 e 4.5).
O uso concomitante de Zanipress com medicamentos contendo aliscireno está contraindicado em doentes com diabetes mellitus ou compromisso renal (TFG <60 ml/min/1,73 m2) (ver secções 4.5 and 5.1).
Hipotensão sintomática:
A hipotensão sintomática é raramente observada em doentes hipertensos sem complicações. Em doentes hipertensos a fazer enalapril, a hipotensão é mais frequentemente observada caso o doente esteja em depleção de volume, i.e., com terapêutica diurética, restrição de sal, diálise, diarreia ou vómitos (ver secção 4.5). Em doentes com insuficiência cardíaca, com ou sem insuficiência renal associada, foi observada hipotensão sintomática. Esta situação tem uma maior probabilidade de ocorrência em doentes num grau mais grave de insuficiência cardíaca, como reflexo de doses elevadas de diuréticos da ansa, hiponatrémia ou disfunção renal. Nestes doentes, a terapêutica deve ser iniciada sob supervisão médica e os doentes devem ser seguidos de perto sempre que a dose de enalapril e/ou diurético é ajustada.
Estas considerações podem ser aplicadas em doentes com isquémia cardíaca ou doença cerebrovascular nos quais uma descida acentuada na pressão arterial pode resultar num enfarte do miocárdio ou acidente vascular cerebral.
Caso ocorra hipotensão, o doente deve ser colocado em posição supina e, se necessário, deve ser-lhe administrado uma perfusão intravenosa de soro fisiológico. Uma resposta hipotensora transitória não é contraindicação para doses mais elevadas, que podem ser administradas normalmente, sem dificuldade, após a pressão arterial ter aumentado após expansão de volume.
Em alguns doentes com insuficiência cardíaca que tenham pressão arterial normal ou baixa, pode ocorrer um abaixamento adicional da pressão arterial sistémica com o enalapril. Este efeito pode ser antecipado e normalmente não é razão para descontinuar o tratamento. Se a hipotensão se tornar sintomática pode ser necessário reduzir a dose e/ou fazer a descontinuação do diurético e/ou enalapril.
Síndrome do nódulo sinusal:
Lercanidipina deve ser administrada com preacução em doentes com síndrome do nódulo sinusal (sem um pacemaker).
Disfunção ventricular esquerda:
Apesar de estudos hemodinâmicos controlados não terem mostrado compromisso da função ventricular, deve ser tomada precaução em doentes com disfunção ventricular esquerda.
Doença Cardíaca:
Isquémica:
Foi sugerido que algumas dihidropiridinas de curta ação podem estar associadas a um risco cardiovascular aumentado em doentes com doença cardíaca isquémica. Apesar de a lercanidipina ser de longa duração, é necessária precaução nestes doentes.
Raramente algumas dihidropiridinas podem causar dor pré-cordial ou angina de peito. Muito raramente, doentes com angina de peito pré-existente podem ter um aumento da frequência, duração ou gravidade dos ataques. Foram observados casos isolados de enfarte do miocárdio (ver secção 4.8).
Compromisso renal:
É necessária precaução com o enalapril no início do tratamento de doentes com compromisso renal leve a moderado. Nestes casos, nos doentes em tratamento com enalapril e por rotina médica, faz-se a monitorização dos níveis de potássio e creatinina.
Foram relatados casos de insuficiência renal associada ao tratamento com enalapril, em particular em doentes com insuficiência cardíaca grave ou com doença renal, incluindo estenose arterial renal.
A insuficiência renal derivada do tratamento com enalapril pode ser reversível se o diagnóstico for feito precocemente e aplicado o tratamento adequado.
Em alguns doentes hipertensos sem doença renal pré-existente, a associação de enalapril com um diurético pode levar a um aumento da urémia e creatinina. Nestes casos, pode ser necessária uma redução da dose do enalapril e/ou descontinuição do diurético. Torna-se necessário o despiste de estenose da artéria renal (ver secção 4.4., Hipertensão arterial renovascular).
Hipertensão arterial renovascular:
Há um aumento do risco de hipotensão ou insuficiência renal nos doentes com estenose renal bilateral ou estenose da artéria de um único rim funcional sob terapêutica de inibidores da ECA. A perda da função renal pode ocorrer com apenas ligeiras alterações na creatinina sérica. Nestes doentes, o tratamento deve ser iniciado sob supervisão médica com doses baixas e titulação cuidadosa e monitorização da função renal.
Transplante renal:
Não existe qualquer experiência na utilização da lercanidipina ou enalapril em doentes que tenham sido sujeitos a transplante renal. Desta forma, o tratamento com Zanipress não é recomendado nestes doentes.
Insuficiência hepática:
O efeito anti-hipertensor da lercanidipina pode ser potenciado em doentes com disfunção hepática.
Muito raramente, o tratamento com inibidores da ECA tem sido associado a um síndroma que tem início com icterícia colestática ou hepatite e progride para necrose hepática fulminante e às vezes morte. O mecanismo deste sindroma não está devidamente explicado. Doentes que desenvolvem icterícia ou uma elevação acentuada das enzimas hepáticas com inibidores da ECA devem parar o tratamento com inibidores da ECA e deve ser-lhes administrado o tratamento adequado.
Diálise Peritoneal:
A lercanidipina foi associada com o desenvolvimento de fluido peritoneal turvo em doentes sujeitos a diálise peritoneal. A turbidez é devida a uma concentração aumentada de triglicéridos no fluido peritoneal. Embora o mecanismo seja desconhecido, a turbidez tem tendência a resolver-se rapidamente após a descontinuação da lercanidipina. Esta é uma associação importante a reconhecer, pois o fluido peritoneal turvo pode ser confundido com peritonite infeciosa com hospitalização consequente desnecessária e administração empírica de antibióticos.
Neutropénia/agranulocitose:
Em doentes com tratamento por inibidores da ECA foram relatados casos de neutropenia/agranulocitose, trombocitopenia e anemia. A neutropenia é rara em doentes com função renal normal e sem fatores de risco particulares. O enalapril deve ser utilizado com elevada precaução em doentes com doença vascular de colagéneo, nos doentes sob tratamento com imunossupressores, alopurinol, procainamida ou se qualquer destes fatores estiver presente, especialmente com compromisso da função renal pré-existente. Ocorreram algumas infeções graves nestes doentes e nalguns casos não houve resposta a tratamento intensivo com antibiótico. Se o enalapril for utilizado nestes doentes, deve ser feita uma monitorização regular de leucócitos e os doentes devem relatar qualquer sinal de infeção ao médico.
Hipersensibilidade/edema angioneurótico:
O edema angioneurótico com envolvimento da face, extremidades, lábios, língua, glote e/ou laringe foi reportado em doentes tratados com inibidores da ECA, incluindo enalapril. Pode ocorrer a qualquer momento do tratamento. Nestes casos, o tratamento com enalapril deve ser imediatamente interrompido e o doente deve ser cuidadosamente monitorizado de forma a assegurar que os sintomas estão totalmente resolvidos antes da saída do hospital. Mesmo nos casos em que ocorre unicamente edema da língua, sem sofrimento respiratório, os doentes podem necessitar de observação prolongada, uma vez que o tratamento com anti- histamínicos e corticosteroides pode não ser suficiente.
Muito raramente, foram relatados casos fatais devido a angioedema associado a um edema da laringe ou edema da língua. Os doentes com envolvimento da língua, glote ou laringe estão mais suscetíveis à obstrução das vias aéreas, especialmente com história de cirurgia das vias respiratórias.
Quando há envolvimento da língua, glote ou laringe, suscetível de causar obstrução respiratória, deve ser instituído de imediato tratamento adequado, o qual poderá incluir a administração subcutânea de uma solução de epinefrina 1:1000 (0,3 ml a 0,5ml) e/ou medidas para assegurar ventilação.
Foi relatada uma incidência superior de casos de angiodema em doentes de raça negra tratados com inibidores da ECA comparados com os de outras raças.
Os doentes com antecedentes de angioedema não despoletado por um inibidor ECA podem estar sujeitos a um risco mais elevado de desenvolverem angioedema se lhes for administrado um inibidor ECA (ver secção 4.3).
O uso concomitante de inibidores da ECA com sacubitril / valsartan é contraindicado devido ao aumento do risco de angioedema. O tratamento com sacubitril / valsartan não deve ser iniciado antes de 36 horas após a última dose de enalapril. O tratamento com enalapril não deve ser iniciado antes de 36 horas após a última dose de sacubitril / valsartan (ver secções 4.3 e 4.5).
O uso concomitante de inibidores da ECA com racecadotril, inibidores da mTOR (por exemplo, sirolimus, everolimus, temsirolimus) e vildagliptina pode levar a um aumento do risco de angioedema (por exemplo, inchaço das vias respiratórias ou língua, com ou sem insuficiência respiratória) (ver secção 4.5). Recomenda-se precaução ao iniciar o racecadotril, os inibidores da mTOR (por exemplo, sirolimus, everolimus, temsirolimus) e a vildagliptina num doente que já esteja a tomar um inibidor da ECA.
Reações Anafilactóides durante a Dessensibilização com Venenos de Insetos Ocorreram raramente reações anafilactóides quase fatais durante a terapia de dessensibilização contravenenos de insetos e uso concomitante de inibidores da ECA. Estas reações podem ser evitadas pela descontinuação de inibidores da ECA antes do início da dessensibilização.
Reações Anafilactóides durante a Aférese LDL:
Ocorreram raramente reações anafilactóides quase fatais durante uma aférese lipoproteica de baixa densidade (LDL) com dextranosulfato e uso simultâneo de inibidores da ECA. Estas reações podem ser evitadas pela descontinuação temporária de inibidores antes de cada aférese.
Hipoglicemia:
Em doentes diabéticos, tratados com antidiabéticos orais ou insulina, que iniciam o tratamento com inibidores da ECA, deve ser feito um controlo cuidadoso da glicemia, especialmente no primeiro mês de tratamento (ver secção 4.5).
Tosse:
O aparecimento de tosse foi associado ao tratamento com inibidores da ECA. Geralmente a tosse é não produtiva, persistente e desaparece após a interrupção do tratamento. A tosse induzida por inibidores da ECA deve ser considerada no diagnóstico diferencial da tosse.
Cirurgia/anestesia:
Em doentes submetidos a cirurgia ou anestesia com agentes que reduzem a pressão arterial, o enalapril inibe a formação da angiotensina II, que normalmente ocorreria devida a uma secreção compensatória de renina. Se a hipotensão for desenvolvida como resultado deste mecanismo, pode ser corrigida através de expansão de volume.
Potássio sérico:
Os inibidores da ECA podem causar hipercaliemia porque inibem a liberação de aldosterona. O efeito geralmente não é significativo em doentes com função renal normal. Contudo, em doentes com insuficiência renal e / ou em doentes a tomar suplementos de potássio (incluindo substitutos do sal), diuréticos poupadores de potássio, trimetoprim ou cotrimoxazol, também conhecidos por sulfametoxazol + trimetoprim e especialmente antagonistas da aldosterona ou bloqueadores do receptor da angiotensina, pode ocorrer hipercaliemia. Os diuréticos poupadores de potássio e os bloqueadores dos recetores da angiotensina devem ser usados com precaução em doentes a receber inibidores da ECA, e o potássio sérico e a função renal devem ser monitorizados (ver secção 4.5).
Lítio:
O uso concomitante de lítio e enalapril, não é geralmente recomendado (ver secção 4.5).
Duplo bloqueio do sistema renina-angiotensina-aldosterona (SRAA):
Há evidência de que o uso concomitante de inibidores da ECA com bloqueadores dos recetores da angiotensina II ou aliscireno aumenta o risco de hipotensão, hipercaliémia e diminuição da função renal (incluindo insuficiência renal aguda). O bloqueio duplo do SRAA, através do uso combinado de um inibidor da ECA e um bloqueador dos recetores da angiotensina II ou aliscireno não é por isso recomendado (ver secções 4.5 e 5.1).
Se a terapêutica de bloqueio duplo for considerada absolutamente necessária, a mesma só deve acontecer sob supervisão de um especialista e sujeita a uma monitorização apertada e frequente da função renal, eletrólitos e pressão arterial.
Os inibidores da ECA e os bloqueadores dos recetores da angiotensina II não devem ser utilizados concomitantemente em doentes com nefropatia diabética.
Indutores de CYP3A4:
Os indutores de CYP3A4 como os anticonvulsivantes (p.ex. fenitoína, carbamazepina) e rifampicina podem reduzir os níveis plasmáticos da lercanidipina pelo que a eficácia da lercanidipina pode ser mais reduzida do que a esperada (ver secção 4.5).
Diferenças étnicas:
Como acontece com outros inibidores da ECA, o enalapril é menos eficaz na redução da pressão arterial em doentes de raça negra comparativamente com os de raça não negra, possivelmente porque os níveis plasmáticos de renina são normalmente mais baixos na população hipertensa de raça negra.
Gravidez:
Zanipress não está recomendado durante a gravidez.
Os inibidores da ECA como o enalapril não devem ser iniciados durante a gravidez. A não ser que a terapêutica com inibidores da ECA seja considerada essencial, as doentes que planeiam engravidar devem mudar para tratamentos alternativos com anti-hipertensores que tenham um perfil de segurança estabelecido para utilização durante a gravidez. Quando a gravidez for diagnosticada, o tratamento com inibidores da ECA deve ser imediatamente interrompido e, se apropriado, deve ser iniciada terapêutica alternativa (ver secções 4.3 e 4.6).
O uso de lercanidipina também não é recomendado durante a gravidez ou em mulheres que planeiam engravidar (ver secção 4.6).
Aleitamento:
Não está recomendado o uso de Zanipress durante o aleitamento (ver secção 4.6).
População pediátrica:
A segurança e eficácia desta associação, não foram demonstradas em crianças.
Álcool:
O álcool deve ser evitado pois pode potenciar o efeito de anti-hipertensores vasodilatadores (ver secção 4.5).
Lactose:
Este mediamento contém lactose. Doentes com problemas hereditários raros de intolerância à galactose, deficiência de lactase ou malabsorção de glucose-galactose não devem tomar este medicamento.
Sódio:
Este medicamento contém menos do que 1 mmol (23 mg) de sódio, por comprimido, ou seja, é praticamente “isento de sódio”.
O efeito anti-hipertensor de Zanipress pode ser potenciado por outros anti- hipertensores tais como diuréticos, b-bloqueantes, a-bloqueantes e outras substâncias.
As seguintes interações foram observadas com um ou outro constituinte da associação:
Maleato de enalapril:
Medicamentos que aumentam o risco de angioedema:
O uso concomitante de inibidores da ECA com sacubitril / valsartan é contraindicado, uma vez que aumenta o risco de angioedema (ver secções 4.3 e 4.4).
O uso concomitante de inibidores da ECA com racecadotril, inibidores da mTOR (por exemplo, sirolimus, everolimus, temsirolimus) e vildagliptina pode levar a um aumento do risco de angioedema (ver secção 4.4).
Duplo bloqueio do sistema renina-angiotensina-aldosterona (SRAA):
Dados de ensaios clínicos têm mostrado que o bloqueio duplo do sistema renina- angiotensina-aldosterona (SRAA) através do uso combinado de inibidores da ECA, bloqueadores dos recetores da angiotensina II ou aliscireno está associado a uma maior frequência de efeitos adversos tais como hipotensão, hipercaliémia e diminuição da função renal (incluindo insuficiência renal aguda), em comparação com a utilização de um único medicamento que atua no SRAA. (ver secções 4.3, 4.4 e 5.1).
Diuréticos poupadores de potássio, suplementos de potássio ou substitutos do sal que contêm potássio:
Embora o potássio sérico geralmente permaneça dentro dos limites normais, pode ocorrer hipercaliemia em alguns pacientes tratados com enalapril. Diuréticos poupadores de potássio (por exemplo, espironolactona, triantereno ou amilorida), suplementos de potássio ou substitutos de sal que contêm potássio podem levar a aumentos significativos no potássio sérico. Cuidados também devem ser tomados quando o enalapril é coadministrado com outros agentes que aumentam o potássio sérico, como trimetoprim e cotrimoxazol (trimetoprim + sulfametoxazol), pois o trimetoprim é conhecido por atuar como um diurético poupador de potássio como a amilorida. Portanto, a combinação de enalapril com os fármacos acima mencionados não é recomendada. Se o uso concomitante for indicado, eles devem ser usados com precaução e com monitorização frequente do potássio sérico.
Ciclosporina:
Pode ocorrer hipercaliemia durante o uso concomitante de inibidores da ECA com ciclosporina. Recomenda-se a monitorização do potássio sérico.
Heparina:
Pode ocorrer hipercaliemia durante o uso concomitante de inibidores da ECA com heparina. Recomenda-se a monitorização do potássio sérico.
Diuréticos (tiazidas ou diuréticos da ansa):
Tratamento prévio com doses elevadas de diuréticos pode levar a uma depleção de volume e risco de hipotensão quando iniciado o tratamento com enalapril (ver secção 4.4). O efeito hipotensor pode ser reduzido pela descontinuação do diurético, pelo aumento do volume ou ingestão de sal, ou iniciando o tratamento com uma dose baixa de enalapril.
Outros anti-hipertensores:
O uso concomitante com outros anti-hipertensores pode aumentar os efeitos hipotensores do enalapril. O uso concomitante de nitroglicerina e outros nitratos ou outros vasodilatadores pode levar a uma redução da pressão arterial.
Lítio:
Foram relatados casos de aumentos reversíveis da concentração de lítio e efeitos tóxicos durante a administração concomitante de lítio com os inibidores da ECA. O uso concomitante de diuréticos tiazidicos pode aumentar as concentrações de lítio e consequentemente aumentar o risco de toxicidade de lítio com os inibidores da ECA. O uso de enalapril com o lítio não é recomendado, mas se a associação for necessária, deve ser feita uma monitorização cuidadosa dos níveis de lítio (ver secção 4.4).
Antidepressivos tricíclicos/ Antipsicóticos/ Anestésicos/ Narcóticos:
O uso concomitante de certos agentes anestésicos, antidepressivos tricíclicos e antipsicóticos com os inibidores da ECA pode resultar numa redução da pressão arterial (ver secção 4.4.).
Anti-inflamatórios Não Esteroides (AINE’s) incluindo Inibidores Seletivos da Cicloxigenase-2 (COX2):
Os Anti-inflamatórios Não Esteroides (AINE’s), incluindo os inibidores seletivos da ciclo-oxigenase 2 (inibidores da COX2) podem reduzir o efeito de diuréticos e outros anti-hipertensores. Por conseguinte, o efeito anti hipertensor dos antagonistas dos recetores da angiotensina II (ARA) ou dos inibidores da ECA podem ser atenuados pelos AINE’s, incluindo os inibidores seletivos da COX2.
A coadministração de AINE’s (incluindo os inibidores da COX2) e inibidores seletivos da ciclo-oxigenase 2 ou inibidores da ECA exercem um efeito aditivo no aumento dos níveis de potássio sérico e podem resultar na deterioração da função renal. Estes efeitos são geralmente reversíveis. Raramente, pode ocorrer insuficiência renal aguda, especialmente em doentes com função renal comprometida (tais como, idosos ou doentes com depleção de volume, incluindo os que fazem tratamento com diuréticos). Desta forma, a associação deve ser administrada com precaução em doentes com a função renal comprometida. Os doentes devem ser adequadamente hidratados e deve-se considerar a monitorização da função renal após o início da terapêutica concomitante e depois periodicamente.
Ouro:
Foram relatadas reações nitritoides raras (os sintomas incluem rubor facial, náuseas, vómito e hipotensão) em terapia com ouro injetável (aurotiomalato de sódio) e terapêutica concomitante com inibidores da ECA incluindo enalapril.
Simpaticomiméticos:
Os simpaticomiméticos podem reduzir o efeito anti hipertensor dos inibidores da ECA.
Antidiabéticos:
Estudos epidemiológicos sugeriram que o uso concomitante de inibidores da ECA e antidiabéticos (insulina, antidiabéticos orais) podem levar a um aumento de redução da glicemia com risco de hipoglicémia. Estes casos são mais suscetíveis de ocorrer nas primeiras semanas de tratamento com a associação e em doentes com compromisso renal (ver secções 4.4 e 4.8).
Álcool:
O álcool aumenta o efeito hipotensor dos inibidores da ECA.
Ácido acetilsalicílico, trombolíticos e ß-bloqueantes:
O enalapril pode ser administrado de forma segura concomitantemente com o ácido acetilsalicílico (em doses adequadas para a profilaxia cardiovascular), trombolíticos e ß-bloqueantes.
Lercanidipina:
Uso concomitante contraindicado.
Inibidores CYP3A4:
A lercanidipina é metabolizada pela enzima CYP3A4e os inibidores do CYP3A4 administrados simultaneamente podem interagir com o metabolismo e eliminação da lercanidipina.
Um estudo de interação com um forte inibidor do CYP3A4, cetoconazol, mostrou um aumento considerável dos níveis plasmáticos da lercanidipina (um aumento de 15 vezes da AUC e um aumento de 8 vezes da Cmax do eutómero S-lercanidipina).
Deve evitar-se a prescrição concomitante de lercanidipina com inibidores do CYP3A4 (p. ex.: cetoconazol, itraconazol, ritonavir, eritromicina, troleandomicina, claritromicina) (ver secção 4.3).
Ciclosporina:
Foram observados aumentos das concentrações plasmáticas de lercanidipina e ciclosporina após a administração concomitante. Um estudo com voluntários jovens saudáveis mostrou que, quando a ciclosporina foi administrada 3 horas após a toma de lercanidipina, os níveis plasmáticos de lercanidipina não sofreram alteração, , no entanto a AUC da ciclosporina aumentou 27%. Contudo, a coadministração de lercanidipina com ciclosporina provocou um aumento de 3 vezes nos níveis plasmáticos de lercanidipina e um aumento de 21% na AUC da ciclosporina.
A ciclosporina e a lercanidipina não devem ser administradas conjuntamente (ver secção 4.3).
Toranja ou sumo de toranja:
Tal como para outras dihidropiridinas, a lercanidipina é sensível à inibidçãodo metabolismo pela toranja ou sumo de toranja, com um aumento consequente na sua disponibilidade sistémica e aumento do efeito hipotensor. A lercanidipina não deve ser administrada com toranja ou sumo de toranja (ver secção 4.3).
Uso concomitante não recomendado:
Indutores do CYP3A4:
A coadministração de lercanidipina com indutores do CYP3A4, tais como anticonvulsivantes (p. ex.: fenitoína, fenobarbital, carbamazepina) e rifampicina deve ser abordada com cuidado, dado que o efeito anti-hipertensor pode ser diminuído e a pressão arterial deve, por isso, ser monitorizada mais frequentemente do que o habitual (ver secção 4.4).
Álcool:
Deve evitar-se o consumo de álcool porque pode potenciar o efeito dos fármacos anti-hipertensores vasodilatadores (ver secção 4.4).
Precauções que incluem ajuste de dose:
Substratos do CYP3A4:
Devem tomar-se cuidados quando a lercanidipina é prescrita concomitantemente com outros substratos do CYP3A4, como terfenadina, astemizol, fármacos antiarrítmicos da classe III, tais como amiodarona, quinidina, sotalol.
Midazolam:
Quando administrada concomitantemente com midazolam a voluntários idosos, numa dose de 20 mg por via oral, a absorção da lercanidipina foi aumentada (em aproximadamente 40%) e a taxa de absorção diminuída (tmax foi retardada de 1,75 para 3 horas). As concentrações de midazolam não sofreram modificações.
Metoprolol:
Quando a lercanidipina foi coadministrada com metoprolol, um beta-bloqueante eliminado principalmente pelo fígado, a biodisponibilidade do metoprolol não foi alterada, enquanto a da lercanidipina foi reduzida em 50%. Este efeito pode dever- se à redução no fluxo sanguíneo hepático causada pelos betabloqueadores e pode, consequentemente, ocorrer com outros fármacos da mesma classe. Consequentemente, a lercanidipina pode ser administrada com segurança conjuntamente com fármacos bloqueadores dos recetores beta-adrenérgicos, mas pode ser necessário um ajuste de dose.
Digoxina:
A coadministração de 20 mg de lercanidipina em doentes submetidos a tratamento crónico com b-metildigoxina não revelou qualquer evidência de interação farmacocinética. No entanto, apresentaram um aumento médio de 33% na Cmax da digoxina, enquanto a AUC e a depuração renal não foram significativamente alteradas. Doentes submetidos a tratamento concomitante com digoxina devem ser cuidadosamente monitorizados clinicamente para sinais de toxicidade pela digoxina.
Uso concomitante com outros medicamentos:
Fluoxetina:
Um estudo de interacção com a fluoxetina (um inibidor do CYP2D6 e CYP3A4), conduzido em voluntários saudáveis com idades de 65 ± 7 anos (média ± d.p.), não demonstrou modificação clinicamente relevante da farmacocinética da lercanidipina.
Cimetidina:
A administração simultânea de 800 mg por dia de cimetidina não causa modificações significativas nos níveis plasmáticos de lercanidipina, porém, em doses mais elevadas, são necessárias precauções, dado que a biodisponibilidade e, consequentemente, o efeito hipotensor da lercanidipina podem ser aumentados.
Sinvastatina:
Quando uma dose de 20 mg de lercanidipina foi reiteradamente coadministrada com 40 mg de sinvastatina, a AUC da lercanidipina não foi significativamente alterada, enquanto a AUC da sinvastatina aumentou 56% e a do seu metabolito ativo, b- hidroxiácido, aumentou 28%. É improvável que tais alterações tenham relevância clínica. Nenhuma interação é esperada quando a lercanidipina for administrada de manhã e a sinvastatina à noite, tal como indicado para este fármaco.
Varfarina:
A coadministração de 20 mg de lercanidipina a voluntários saudáveis, em jejum, não alterou a farmacocinética da varfarina.
Diuréticos e Inibidores da ECA:
A lercanidipina foi administrada com segurança com diuréticos e inibidores da ECA.
Outros medicamentos que afetam a pressão arterial:
Como para todos os medicamentos anti-hipertensores, um efeito hipotensor aumentado pode ser observado quando a lercanidipina é administrada com outros medicamentos que afetam a pressão arterial, como os bloqueadores alfa para o tratamento de sintomas urinários, antidepressivos tricíclicos, neurolépticos. Ao contrário, uma redução do efeito hipotensor pode ser observada com o uso concomitante de corticosteroides.
População pediátrica:
Os estudos de interação só foram realizados em adultos.
Gravidez:
Enalapril:
A utilização de inibidores da ECA (enalapril) não é recomendada durante o primeiro trimestre de gravidez (ver secção 4.4). A utilização de inibidores da ECA (enalapril) está contraindicada durante o segundo e terceiro trimestres de gravidez (ver secções 4.3 e 4.4).
A evidência epidemiológica relativa ao risco de teratogenicidade após exposição aos inibidores da ECA durante o primeiro trimestre da gravidez não foi conclusiva; no entanto, um pequeno aumento do risco não pode ser excluído. A não ser que a terapêutica continuada com inibidores da ECA seja considerada essencial, as doentes que planeiam engravidar devem substituir o tratamento por terapêuticas anti hipertensoras que tenham um perfil de segurança bem estabelecido para utilização na gravidez. Quando a gravidez for diagnosticada, o tratamento com inibidores da ECA deve ser imediatamente interrompido e, se apropriado, deve ser iniciada uma terapêutica alternativa.
A exposição aos inibidores da ECA durante o segundo e terceiro trimestres induz fetotoxicidade humana (função renal diminuída, oligohidrâmnios, atraso da ossificação do crânio) e toxicidade neonatal (insuficiência renal, hipotensão, hipercaliémia) (ver secção 5.3). Ocorreu oligoidrâmnios materno, presumivelmente representando diminuição da função renal fetal, o qual pode resultar em contraturas dos membros, deformações craniofaciais e desenvolvimento de pulmão hipoplásico. No caso de ter havido exposição aos inibidores da ECA a partir do segundo trimestre de gravidez, recomendam-se exames de ultrassonografia à função renal e ao crânio. Os recém-nascidos cujas mães tenham tomado inibidores da ECA devem ser cuidadosamente observados relativamente a hipotensão (ver secções 4.3 e 4.4).
Lercanidipina:
Não estão disponíveis dados sobre o uso de lercanidipina em mulheres grávidas. Estudos em animais não demonstraram efeitos teratogénicos (ver secção 5.3), mas estes têm sido observados com outros compostos dihidropiridinicos.
A lercanidipina não está recomendada durante a gravidez e em mulheres em idade fértil, que não utilizammétodos contracetivos (ver secção 4.4).
Lercanidipina e enalapril em associação:
Não há ou há uma quantidade limitada de dados sobre a utilização de maleato de enalapril + cloridrato de lercanidipina em mulheres grávidas. Os estudos em animais são insuficientes no que respeita à toxicidade reprodutiva (ver secção 5.3).
Zanipress não deve ser utilizado no segundo e terceiro trimestres de gravidez. Não é recomendado no primeiro trimestre de gravidez e em mulheres em idade fértil, que não utilizam métodos contracetivos.
Amamentação:
Enalapril:
Os dados farmacocinéticos são limitados e demonstraram concentrações muito baixas no leite materno (ver secção 5.2). Embora estas concentrações pareçam ser clinicamente irrelevantes, a utilização de Enalapril durante o aleitamento não está recomendada para recém-nascidos prematuros e durante as primeiras semanas após o parto, devido ao risco hipotético de efeitos cardiovasculares e renais, e porque não há suficiente experiência clínica. No caso de uma criança mais velha, a utilização de enalapril pode ser considerada na mãe a amamentar, se este tratamento for necessário para a mãe e os efeitos adversos forem monitorizados na criança.
Lercanidipina:
Desconhece-se se a lercanidipina / metabolitos são excretados no leite humano. Não pode ser excluído um risco para os recém-nascidos / lactentes. A lercanidipina não deve ser utilizada durante a amamentação.
Lercanidipina e enalapril em associação:
Consequentemente, não está recomendada a utilização de Zanipress durante o aleitamento.
Fertilidade:
Não existem dados clínicos disponiveis com lercanidipina. Foram relatados alguns casos de alterações bioquímicas reversíveis na cabeça dos espermatozoides, que podem prejudicar a fecundação, em doentes tratados com bloqueadores de cálcio. Nos casos de insucesso repetido de fertilizações in vitro e em que nenhuma outra explicação é encontrada, deve ser considerada como causa a utilização de bloquedores dos canais de cálcio.
Zanipress tem pouca influência sobre a capacidade de conduzir e utilizar máquinas. No entanto, deve ter-se precaução, pois podem ocorrer tonturas, astenia, fadiga e em casos raros sonolência. (ver secção 4.8).
Resumo do perfil de segurança:
A segurança de Zanipress foi avaliada em cinco ensaios clínicos controlados, duplo- cegos, e em duas fases de extensão aberta de longo prazo. No total, 1.141 doentes receberam uma dose de 10 mg + 10 mg, 20 mg + 10 mg e 20 mg + 20 mg de Zanipress. Os efeitos indesejáveis observados com a associação são semelhantes aos observados individualmente para cada constituinte. As reações adversas mais frequentemente relatadas durante o tratamento com Zanipress foram tosse (4,03%), tontura (1,67%) e cefaleia (1,67%).
Tabela sumária das reações adversas:
A tabela seguinte apresenta as reações adversas relatadas em ensaios clínicos com Zanipress 10 mg + 10 mg, 20 mg + 10 mg e 20 mg + 20 mg e para as quais existe uma relação causal razoável, agrupadas por Classes de Sistemas de Órgãos segundo a classificação MedDRA, e ordenadas por frequência: muito frequentes (> 1/10), frequentes (≥ 1/100 a <1/10), pouco frequentes (≥ 1/1000 a <1/100), raros (≥ 1/10 000, <1/1, 000), muito raros (<1/10 000) desconhecida (não pode ser estimada a partir dos dados disponíveis).
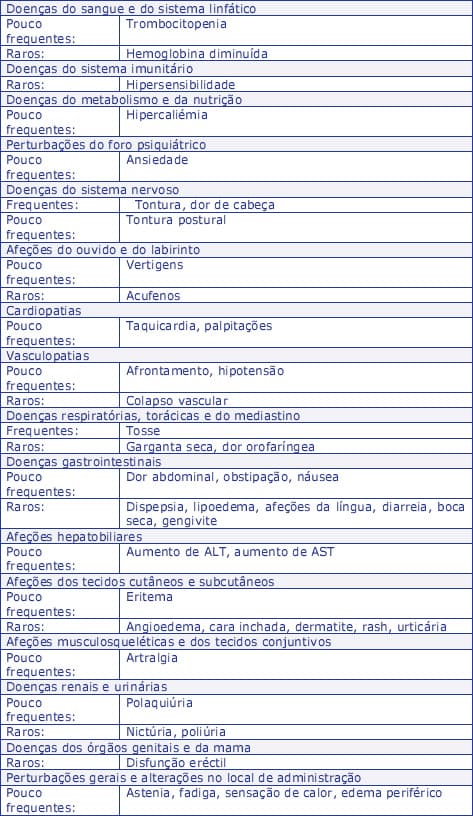
Os efeitos indesejáveis que ocorrem apenas num doente são classificados como raros.
Descrição das reações adversas selecionadas:
A incidência de reações adversas selecionadas, frequentemente observadas com enalapril e lercanidipina em monoterapia, é mostrada na tabela abaixo, conforme relatado num ensaio clínico fatorial, aleatorizado, duplamente-cego:
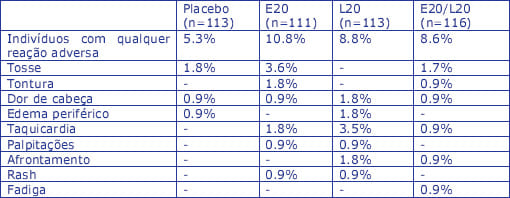
Informação adicional dos componentes individuais:
As reações adversas notificadas com um dos componentes individuais (enalapril ou lercanidipina) podem ser também potenciais efeitos indesejáveis com Zanipress, mesmo se não forem observadas nos ensaios clínicos ou durante o período pós- comercialização.
Enalapril individualmente:
De entre as reações adversas relatadas para enalapril estão:
Doenças do sangue e do sistema linfático:
Pouco frequentes: anemia (incluindo aplástica e forma hemolítica).
Raros: neutropenia, diminuições na hemoglobina, diminuições no hematócrito, trombocitopenia, agranulocitose, depressão da medula óssea, pancitopenia, linfadenopatia, doenças auto-imunes.
Doenças endócrinas:
Desconhecidos: síndrome de secreção inadequada da hormona antidiurética (SIADH).
Doenças do metabolismo e da nutrição:
Pouco frequentes: hipoglicémia (ver secção 4.4.).
Perturbações do foro psiquiátrico:
Frequentes: depressão.
Pouco frequentes: confusão, nervosismo, insónia.
Raros: sonhos anormais, distúrbios do sono.
Doenças do sistema nervoso:
Muito frequentes: tonturas.
Frequentes: dor de cabeça, síncope, alterações do paladar.
Pouco frequentes: sonolência, parestesia, vertigens.
Afeções oculares:
Muito frequentes: visão turva.
Afeções do ouvido e do labirinto:
Pouco frequentes: acufenos.
Cardiopatias:
Frequentes: dor no peito, alterações do ritmo, angina de peito, taquicardia.
Pouco frequentes: palpitações, enfarte do miocárdio ou acidente cerebrovascular*, possivelmente secundário a hipotensão excessiva em doentes de alto risco (ver secção 4.4.).
* As taxas de incidência foram comparáveis às dos grupos placebo e controlo ativo nos ensaios clínicos
Vasculopatias:
Frequentes: hipotensão (incluindo hipotensão ortostática).
Pouco frequentes: rubor, hipotensão ortostática.
Raros: fenómeno de Raynaud.
Doenças respiratórias, torácicas e do mediastino:
Muito frequentes: tosse.
Frequentes: dispneia.
Pouco frequentes: rinorreia, faringite e rouquidão, broncoespasmo/asma.
Raros: infiltração pulmonar, rinite, alveolite alérgica/pneumonia eosinofílica.
Doenças gastrointestinais:
Muito frequentes: náusea.
Frequentes: diarreia, dor abdominal.
Pouco frequentes: íleo, pancreatite, vómitos, dispepsia, obstipação, anorexia, irritação gástrica, boca seca, úlcera péptica.
Raros: estomatite/estomatite aftosa, glossite.
Muito raros: angioedema intestinal.
Afeções hepatobiliares:
Raros: insuficiência hepática, hepatite – hepatocelular ou colestática, hepatite incluíndo necrose, colestase (incluindo icterícia).
Afeções dos tecidos cutâneos e subcutâneos:
Frequentes: rash, hipersensibilidade/edema angioneurótico: foram notificados edema angioneurótico da face, extremidades, lábios, língua, glote e/ou laringe (ver secção 4.4).
Pouco frequentes: hipersudação, prurido, urticária, alopécia.
Raros: eritema multiforme, síndroma de Stevens-Johnson, dermatite exfoliativa, necrólise epidérmica tóxica, pênfigo, eritrodermite.
Foi relatado um sintoma complexo que pode incluir alguns ou todos dos seguintes sintomas: febre, serosite, vasculite, mialgia/miosite, artralgia/artrite, anticorpos antinucleares positivos (ANA), aumento da taxa de sedimentação eritrocitária, eosinofilia e leucocitose. Pode ocorrer rash, fotossensibilidade ou outras manifestações dermatológicas.
Afeções musculosqueléticas e dos tecidos conjuntivos:
Pouco frequentes: cãibras musculares.
Doenças renais e urinárias:
Pouco frequentes: disfunção renal, insuficiência renal, proteinúria.
Raros: oligúria.
Doenças dos órgãos genitais e da mama:
Pouco frequentes: impotência.
Raros: ginecomastia.
Perturbações gerais e alterações no local de administração:
Muito frequentes: astenia.
Frequentes: fadiga.
Pouco frequentes: mal-estar, febre.
Exames complementares de diagnóstico:
Frequentes: hipercaliémia, aumento da creatininémia.
Pouco frequentes: aumento da uremia, hiponatremia.
Raros: aumento das enzimas hepáticas, aumento da bilibirrubina sanguínea.
Lercanidipina individualmente:
As reações adversas mais frequentemente notificadas em ensaios clínicos e na experiência pós-comercialização são edema periférico, dores de cabeça, rubor, taquicardia e palpitações.
Doenças do sistema imunitário:
Raros: hipersensibilidade.
Doenças do sistema nervoso:
Frequentes: dor de cabeça.
Pouco frequentes: tonturas.
Raros: sonolência, síncope.
Cardiopatias:
Frequentes: taquicardia, palpitações.
Raros: angina de peito.
Vasculopatias:
Frequentes: rubor.
Pouco frequentes: hipotensão.
Doenças gastrointestinais:
Pouco frequentes: náusea, dispepsia, dor abdominal superior.
Raros: vómitos, diarreia.
Desconhecidos: hipertrofia gengival*, fluído peritoneal turvo*.
Afeções hepatobiliares:
Desconhecidos: transaminases séricas aumentadas*.
Afeções dos tecidos cutâneos e subcutâneos:
Pouco frequentes: rash, prurido.
Raros: urticária.
Desconhecidos: angioedema*.
Afeções musculosqueléticas e dos tecidos conjuntivos:
Pouco frequentes: mialgia.
Doenças renais e urinárias:
Pouco frequentes: poliúria.
Raros: polaquiúria.
Perturbações gerais e alterações do local de administração:
Frequentes: edema periférico.
Pouco frequentes: astenia, fadiga.
Raros: dores no peito.
* Reações adversas provenientes de notificações espontâneas em experiência pós- comercialização mundial.
Algumas dihidropiridinas podem raramente levar a dor precordial ou angina de peito. Muito raramente, doentes com angina de peito pré-existente podem ter um aumento da frequência, duração ou gravidade das crises anginosas. Podem ser observados casos isolados de enfarte do miocárdio.
A lercanidipina não parece estar relacionada com qualquer efeito adverso a nível da glicémia ou dos níveis lipídicos séricos.
Notificação de suspeitas de reações adversas:
A notificação de suspeitas de reações adversas após a autorização do medicamento é importante, uma vez que permite uma monitorização contínua da relação benefício- risco do medicamento. Pede-se aos profissionais de saúde que notifiquem quaisquer suspeitas de reações adversas através do sistema nacional de notificação mencionado abaixo:
Sítio da internet: http://www.infarmed.pt/web/infarmed/submissaoram (preferencialmente) ou através dos seguintes contactos:
Direção de Gestão do Risco de Medicamentos
Parque da Saúde de Lisboa, Av. Brasil 53
1749-004 Lisboa
Tel: +351 21 798 73 73
Linha do Medicamento: 800222444 (gratuita)
E-mail: farmacovigilancia@infarmed.pt
Na experiência pós-comercialização, foram notificados alguns casos de sobredosagem intencional requerendo hospitalização, com a administração de enalapril/lercanidipina em doses de 100 a 1000 mg cada. Os sintomas notificados (pressão arterial sistólica diminuída, bradicardia, agitação, sonolência e dor do flanco), também podem dever-se à administração concomitante de doses elevadas de outros fármacos (por exemplo, betabloqueadores).
Sintomas de sobredosagem com enalapril e lercanidipina individualmente:
Os sintomas mais relevantes de sobredosagem com enalapril relatados são hipotensão acentuada (com início cerca de seis horas após a ingestão dos comprimidos), concomitantemente com o bloqueio do sistema renina-angiotensina e estupor. Os sintomas associados à sobredosagem dos inibidores da ECA podem incluir choque circulatório, distúrbios eletrolíticos, insuficiência renal, hiperventilação, taquicardia, palpitações, bradicardia, tonturas, ansiedade e tosse. Foram relatados níveis séricos de enalaprilato 100 e 200 vezes mais elevados do que os observados habitualmente após doses terapêuticas, após a ingestão de 300 mg e 440 mg de enalapril, respetivamente.
Como com outras dihidropiridinas, a sobredosagem com lercanidipina resulta na vasodilatação periférica excessiva com hipotensão marcada e taquicardia reflexa. No entanto, em doses muito altas, a seletividade periférica pode ser perdida, causando bradicardia e um efeito inotrópico negativo. As reações adversas mais frequentes associadas a casos de sobredosagem foram hipotensão, tonturas, dores de cabeça e palpitações.
Tratamento das situações de sobredosagem com enalapril e lercanidipina individualmente:
O tratamento recomendado na sobredosagem com enalapril é a perfusão intravenosa de solução salina. Se ocorrer hipotensão, o doente deverá ser colocado na posição de choque. Se disponível, o tratamento com perfusão de angiotensina II e/ou catecolaminas intravenosas pode ser também considerado. Se os comprimidos foram ingeridos recentemente, devem ser tomadas medidas para eliminar o maleato de enalapril (p.ex: vómito, lavagem gástrica, administração de adsorventes ou sulfato de sódio). O enalaprilato pode ser removido da circulação através de hemodiálise (ver secção 4.4). A terapêutica com pacemaker está indicada em casos de bradicardia resistente à terapêutica. Sinais vitais, eletrólitos séricos e concentrações de creatinina devem ser continuamente monitorizados.
Com a lercanidipina, a hipotensão clinicamente significativa, requer um suporte cardiovascular ativo, incluindo monitorização frequente da função cardíaca e respiratória, elevação das extremidades e atenção ao volume de fluido circulante e de urina. Tendo em consideração o efeito farmacológico prolongado da lercanidipina, é essencial que o estado cardiovascular dos doentes seja monitorizado durante pelo menos 24 horas. Uma vez que o produto tem uma alta ligação às proteínas, a diálise provavelmente não será eficaz. Os doentes para os quais se antecipe uma intoxicação moderada a grave devem ser observados num ambiente de cuidados intensivos.
Grupo farmacoterapêutico: 3.4.2.1 Inibidores da enzima de conversão da angiotensina, 3.4.3 Bloqueadores da entrada do cálcio, Código ATC: C09BB02.
Zanipress é uma associação fixa de um inibidor ECA (enalapril) e um bloqueador dos canais de cálcio (lercanidipina), dois agentes anti-hipertensores com mecanismos de ação complementares para controlar a pressão arterial em doentes com hipertensão essencial.
Enalapril:
O maleato de enalapril é o sal de enalapril, um derivado de dois aminoácidos, L- alanina e L-prolina. A enzima de conversão da angiotensina (ECA) é uma peptidil dipeptidase que cataliza a conversão da angiotensina I no agente vasoconstritor angiotensina II. Após a absorção, o enalapril é hidrolisado a enaprilato que inibe a ECA. A inibição da ECA resulta num decréscimo plasmático da angiotensina II, o que leva a um aumento plasmático da atividade da renina (devido à retirada do efeito feedback negativo da libertação da renina) e decréscimo da secreção da aldosterona. Uma vez que a ECA é idêntica à cinase II, o enalapril pode também inibir a degradação da bradiquinina, um potente péptido vasoconstritor. No entanto, o papel deste mecanismo nos efeitos terapêuticos do enalapril ainda não está esclarecido.
Apesar do mecanismo pelo qual o enalapril reduz a pressão arterial ser primariamente atribuído à supressão do sistema renina-angiotensina-aldosterona, o enalapril é um anti hipertensor mesmo em doentes com baixos níveis de renina.
A administração de enalapril a doentes hipertensos reduz tanto a pressão arterial supina como a de posição levantada, sem um aumento significativo da frequência cardíaca.
A hipotensão postural sintomática é rara. Em alguns doentes, o controlo ótimo da pressão arterial pode levar algumas semanas de tratamento. A interrupção abrupta de enalapril não está associada a um aumento rápido da pressão arterial.
A inibição efetiva da atividade ECA ocorre normalmente 2 a 4 horas após a administração oral de uma dose única de enalapril. O início de efeito da ação anti hipertensora foi observado após uma hora, com um máximo de redução da pressão arterial observado 4 a 6 horas após a administração. A duração de ação encontra-se relacionada com a dose, mas nas doses recomendadas, os efeitos anti hipertensor e hemodinâmico persistiram pelo menos durante 24 horas.
Em estudos hemodinâmicos em doentes com hipertensão essencial, a redução da pressão arterial estava associada a uma diminuição da resistência arterial periférica com um aumento no débito cardíaco e pouca ou nenhuma alteração na frequência cardíaca. Após a administração de enalapril, ocorreu um aumento no fluxo sanguíneo renal; a taxa de filtração glomerular permaneceu igual. Não houve sinais de retenção de sódio ou de água. No entanto, em doentes com uma taxa de filtração glomerular baixa antes do início do tratamento, as taxas geralmente aumentaram.
Em ensaios clínicos de curta duração em doentes diabéticos e não diabéticos com doença renal, foram observadas diminuição da albuminúria e excreção renal de IgG e proteínas totais, após a administração de enalapril.
Dois ensaios controlados e aleatorizados, o ONTARGET (ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial) e o VA NEPHRON-D (The Veterans Affairs Nephropathy in Diabetes) avaliaram o uso da associação de um inibidor da ECA com um bloqueador dos recetores da angiotensina II.
O ONTARGET foi um estudo conduzido em doentes com história de doença cardiovascular ou cerebrovascular, ou diabetes mellitus tipo 2 acompanhada por evidência de lesão nos órgãos-alvo. O VA NEPHRON-D foi um estudo em doentes com diabetes mellitus tipo 2 e nefropatia diabética.
Estes estudos não demonstraram qualquer efeito benéfico significativo nos resultados da função renal e/ou cardiovascular e mortalidade, apesar de ter sido observado um aumento do risco de hipercaliémia, lesão renal aguda e/ou hipotensão, em comparação com a monoterapia. Dadas as suas propriedades farmacodinâmicas semelhantes, estes resultados também são relevantes para outros inibidores da ECA e bloqueadores dos recetores da angiotensina II.
Os inibidores da ECA e os bloqueadores dos recetores da angiotensina II não devem, portanto, ser utilizados concomitantemente em doentes com nefropatia diabética.
O ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) foi um estudo desenhado para avaliar o benefício da adição de aliscireno a uma terapêutica standard de um inibidor da ECA ou um bloqueador dos recetores da angiotensina II em doentes com diabetes mellitus tipo 2 e doença renal crónica, doença cardiovascular, ou ambos. O estudo foi interrompido precocemente devido a um aumento do risco de efeitos adversos. Morte cardiovascular e acidente vascular cerebral foram ambos numericamente mais frequentes no grupo aliscireno do que no grupo placebo e os efeitos adversos e efeitos adversos graves de interesse (hipercaliemia, hipotensão e disfunção renal) foram notificadas mais frequentemente no grupo de aliscireno do que no grupo placebo.
Lercanidipina:
A lercanidipina é um antagonista do cálcio do grupo das dihidropiridinas e inibe o influxo transmembranar do cálcio para o músculo liso e cardíaco. O mecanismo de ação antihipertensivo é baseado num efeito relaxante direto no músculo liso vascular, levando a uma redução da resistência periférica total. Apesar da semivida plasmática farmacocinética curta, a lercanidipina tem uma ação antihipertensiva prolongada devido ao seu grande coeficiente de partição membranar e não apresenta efeitos inotrópicos negativos devido à sua elevada seletividade vascular.
Uma vez que a vasodilatação produzida pela lercanidipina tem um início de ação gradual, raramente se tem observado hipotensão aguda com taquicardia reflexa nos doentes hipertensos.
Tal como com outras 1,4-dihidropiridinas assimétricas, a atividade anti-hipertensora da lercanidipina deve-se principalmente ao S-enantiómero.
Enalapril + Lercanidipina:
A associação destas substâncias tem um efeito anti-hipertensor aditivo, reduzindo a pressão arterial a um grau maior do que cada substância isolada.
• Zanipress 10mg + 10mg:
Num ensaio clínico piloto de fase III, duplamente cego, conduzido em 342 doentes sem resposta à lercanidipina 10 mg (definidos como TAD 95-114 e TAS 140-189 mmHg) a redução na TAS foi 5,4 mmHg superior com a combinação de enalapril 10mg + lercanidipina 10 mg do que com lercanidipina 10 mg em monoterapia, após 12 semanas de tratamento duplamente cego (-7,7 mmHg vs -2,3 mmHg, p < 0,001). Também a redução na TAD foi 2,8 mmHg superior com a associação comparativamente com a monoterapia (-7,1 mmHg vs -4,3 mmHg, p < 0,001).
As taxas de resposta apresentaram-se significativamente mais elevadas com a terapia de associação do que com a monoterapia: 41% vs 24% (p < 0,001) para a TAS e 35% vs 24% (p= 0,032) para a TAD. Uma percentagem significativamente maior de doentes a fazer a associação teve normalização da TAS (39% vs 22%, p < 0,001) e da TAD (29% vs 19%, p= 0,023) comparativamente com os doentes em monoterapia. Na fase aberta de follow up do estudo foi permitida uma titulação da associação enalapril 20 mg + lercanidipina 10 mg se a PA permanecesse> 140/90mmHg; a titulação ocorreu em 133/221 e a TAD normalizou após titulação em 1/3 dos casos.
• Zanipress 20mg + 10mg:
Num ensaio clínico piloto de fase III, duplamente cego, conduzido em 327 doentes sem resposta ao enalapril 20 mg (definidos como TAD 95-114 e TAS 140-189 mmHg), os doentes que fizeram terapêutica com enalapril 20 mg + lercanidipina 10 mg alcançaram uma redução mais significativa da TAS comparativamente com aqueles em monoterapia (-9,8 vs -6,7 mmHg, p= 0,013) e na TAD (-9,2 vs -7,5 mmHg, p= 0,015). As taxas de resposta não foram significativamente mais elevadas com a associação do que em monoterapia (53% vs 43%, p= 0,076 para a TAD e 41% vs 33%, p= 0,116 para a TAS) e uma percentagem não significativamente mais elevada de doentes na associação apresentou normalização da TAD (48% vs 37%, p= 0,055) e da TAS (33% vs 28%, p= 0,325) comparativamente com doentes em monoterapia.
• Zanipress 20mg + 20mg:
Num ensaio conduzido em dupla ocultação, aleatorizado, contra placebo e controlo- ativo, com um desenho fatorial, conduzido em 1.039 doentes com hipertensão moderada (definidos como TAS 100-109 mmHg, TAD <180 mmHg e TAD em ambulatório ≥ 85 mmHg), os doentes que fizeram terapêutica com enalapril 20mg + lercanidipina 20 mg tiveram reduções mais significativas da TA, quer no consultório quer em ambulatório, medida na posição de sentado, em comparação com o placebo (p <0,001). Foram observadas diferenças clinicamente relevantes na alteração versus a medição inicial da TA diastólica, na posição de sentado no consultório, no vale entre a terapêutica com a associação 20mg + 20mg (-15,2 mmHg, n= 113), em comparação com 20 mg de enalapril (-11,3 mm Hg, p= 0,004, n= 113) ou 20 mg de lercanidipina isolados (-13,0 mm Hg, p= 0,092, n= 113). Da mesma forma, foram observadas diferenças clinicamente relevantes na alteração versus a medição inicial da TA sistólica, na posição de sentado no consultório, no vale entre a terapêutica com a associação 20mg + 20mg (-19,2 mmHg) em comparação com a lercanidipina 20 mg (-13,0 mm Hg, p= 0,002) ou enalapril 20 mg isolados (-15,3 mmHg, p=0,055). Também foram observadas diferenças clinicamente relevantes nas TA sistólica e diastólica em ambulatório. Observou-se um aumento significativo das taxas de resposta para a TAD (75%) e TAS (71%) na terapêutica com a associação 20mg + 20mg em relação ao placebo (p < 0,001) e ambas as monoterapias (p < 0,01). A normalização da pressão arterial foi obtida por uma maior percentagem de doentes tratados com a associação 20mg + 20mg (42 %) do que com o placebo (22%).
Não foram observadas interações farmacocinéticas na associação de enalapril com lercanidipina.
Farmacocinética de enalapril:
Absorção:
O enalapril oral é rapidamente absorvido, com o pico de concentrações plasmáticas de enalapril a ocorrer numa hora. Com base na recuperação urinária, a extensão da absorção de enalapril a partir do maleato de enalapril oral é de 60%. A absorção oral de enalapril não é afetada pela presença de alimentos no trato gastrointestinal.
Distribuição:
Após a absorção, o enalapril oral é rápida e extensamente hidrolisado a enaprilato, um potente inibidor da enzima de conversão da angiotensina. Os picos de concentração do enaprilato ocorrem cerca de 4 horas após a administração oral do maleato de enalapril. A semivida efetiva para acumulação de enalaprilato após múltiplas doses de enalapril oral é de 11 horas. Em doentes com função renal normal, as concentrações séricas em steady-state (equilíbrio) de enaprilato foi atingida após 4 dias de tratamento.
Entre o intervalo de concentrações que são terapeuticamente relevantes, a ligação do enalaprilato às proteínas plasmáticas humanas não ultrapassa os 60%.
Biotransformação:
Para além da conversão em enaprilato, não existe evidência de metabolização significativa do enalapril.
Eliminação:
A eliminação de enaprilato é principalmente renal. Os principais componentes na urina são o enaprilato, cerca de 40% da dose, e enalapril não modificado (cerca de 20%).
Compromisso renal:
A exposição ao enalapril e enaprilato está aumentada em doentes com insuficiência renal. Em doentes com insuficiência renal ligeira a moderada (clearance da creatinina 40-60 ml/min), o steady state AUC do enaprilato foi aproximadamente duas vezes superior do que em doentes com função renal normal após administração de 5 mg uma vez ao dia. Em situações de insuficiência renal grave (clearance da creatinina £ 30 ml/min) a AUC aumentou cerca de 8 vezes. A semivida efetiva do enaprilato a seguir a doses repetidas de maleato de enalapril é prolongada a este nível de insuficiência renal e o tempo para atingir o steady state aumenta (ver secção 4.2).
O enaprilato pode ser removido da circulação geral por hemodiálise. A clearance renal é 62 ml/min.
Aleitamento:
Após uma dose oral única de 20 mg em cada cinco mulheres no pós-parto, a média da concentração máxima de enalapril no leite foi 1.7 µg/L (intervalo 0,54-5,9 mg/L) 4 a 6 horas após a administração. A média da concentração máxima de enaprilato foi 1.7 µg/L (intervalo de 1,2 a 2.3 µg/L); os picos ocorreram em vários momentos durante o período de 24 horas. Utilizando os dados das concentrações máximas no leite, estima-se que a ingestão máxima de enalapril por um bebé alimentado exclusivamente com leite materno seja de cerca de 0,16% da dose materna ajustada ao peso. Uma mulher que esteve a tomar diariamente 10 mg de enalapril por via oral, durante 11 meses atingiu concentrações máximas de enalapril no leite de 2 µg/L, 4 horas após a administração e de enaprilato de 0,75 mg/L, cerca de 9 horas após a administração. A quantidade total de enalapril e enaprilato medidos no leite durante o período de 24 horas foi de 1.44 µg/L e 0,63 µg/L, respetivamente. Os níveis de enaprilato no leite foram indetetáveis 4 horas após a administração de uma dose única de 5 mg de enalapril (<0.2 µg/L) numa mãe e de uma dose única de 10 mg em duas mães; os níveis de enalapril não foram determinados.
Farmacocinética da lercanidipina:
Absorção:
A lercanidipina é completamente absorvida após administração oral e os níveis plasmáticos máximos, ocorrem cerca de 1,5 – 3 horas após a administração.
Os dois enantiómeros de lercanidipina apresentam um perfil de nível plasmático similar: o tempo para se atingir a concentração plasmática máxima é o mesmo, a concentração plasmática máxima e a AUC são, em média, 1,2 vezes mais elevadas para o (S)-enantiómero. As semividas de eliminação dos dois enantiómeros são essencialmente as mesmas. Não se observou interconversão dos enantiómeros in vivo.
Devido a um elevado metabolismo de primeira passagem, a biodisponibilidade absoluta da lercanidipina administrada por via oral a doentes em condições de ausência de jejum, é de cerca de 10%. No entanto, quando administrada a voluntários saudáveis, em jejum, a biodisponibilidade é reduzida para 1/3.
A disponibilidade oral da lercanidipina aumenta 4 vezes quando é ingerida até 2 horas após uma refeição rica em gorduras. Assim, a lercanidipina deve ser tomada antes das refeições.
Distribuição:
A distribuição do plasma para os tecidos e órgãos é rápida e extensa.
O grau de ligação da lercanidipina às proteínas plasmáticas é superior a 98%. Uma vez que os níveis de proteínas plasmáticas estão reduzidos em doentes com disfunção hepática ou renal grave, a fração livre do fármaco pode estar aumentada.
Biotransformação:
A lercanidipina é extensamente metabolizada pelo CYP3A4; não se encontra o fármaco precursor na urina ou nas fezes. É convertida predominantemente em metabolitos inativos e aproximadamente 50% da dose é excretada na urina.
Testes “in vitro” com microssomas do fígado humano demonstraram que a lercanidipina apresenta um certo grau de inibição do CYP3A4 e do CYP2D6, em concentrações 160 e 40 vezes superiores, respetivamente, às alcançadas no pico máximo plasmático após a administração de uma dose de 20 mg.
Além disso, estudos de interação em seres humanos mostraram que a lercanidipina não alterou os níveis plasmáticos de midazolam, um substrato típico do CYP3A4, nem do metoprolol, um substrato típico do CYP2D6. Consequentemente, não é previsível a inibição da biotransformação de fármacos metabolizados pelo CYP3A4 e CYP2D6 com doses terapêuticas de lercanidipina.
Eliminação:
A eliminação processa-se essencialmente por biotransformação.
Foi calculada uma semivida média de eliminação terminal de 8-10 horas e devido à sua elevada ligação aos lípidos da membrana, a atividade terapêutica dura 24 horas. Não se observou acumulação durante a administração repetida.
Linearidade/não linearidade:
A administração oral de lercanidipina origina níveis plasmáticos de lercanidipina que não são diretamente proporcionais à dose (cinética não linear). Após 10, 20 ou 40 mg, as concentrações plasmáticas máximas encontravam-se num rácio de 1:3:8 e as áreas sob as curvas de concentração plasmática-tempo no rácio de 1:4:18, sugerindo uma saturação progressiva do metabolismo de primeira passagem. Em conformidade, a disponibilidade aumenta com a elevação da dose.
Populações especiais:
Em doentes idosos e em doentes com insuficiência renal ligeira a moderada ou insuficiência hepática ligeira a moderada, verificou-se que o comportamento farmacocinético da lercanidipina é similar ao observado na população de doentes em geral; doentes com insuficiência renal grave ou doentes dependentes de diálise apresentaram concentrações mais elevadas do fármaco (cerca de 70%). Em doentes com insuficiência hepática moderada a grave, é provável que a biodisponibilidade sistémica da lercanidipina seja aumentada, dado que, normalmente, o fármaco é extensamente metabolizado no fígado.
Associação enalapril + lercanidipina:
A toxicidade potencial da combinação fixa de enalapril e lercanidipina foi estudada em ratos após a administração oral até 3 meses e com dois testes de genotoxicidade. A associação não alterou o perfil toxicológico dos dois componentes individuais.
A seguinte informação existe para cada um dos componentes individuais, enalapril e lercanidipina.
Enalapril:
Os dados não clínicos não revelaram riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade e potencial carcinogénico.
Os estudos de toxicidade reprodutiva sugerem que o enalapril não tem efeitos na fertilidade e capacidade reprodutiva em ratos e não é teratogénico. Num estudo, no qual, ratos fêmea foram tratados antes da conceção e durante a gestação ocorreu um aumento na incidência de mortes das crias durante a lactação. O composto demonstrou atravessar a placenta e ser excretado no leite. Os inibidores da enzima de conversão da angiotensina, como classe, demonstraram induzir efeitos adversos na fase final do desenvolvimento fetal, resultando em mortes fetais e efeitos congénitos, principalmente a afetar o crânio. Foram também relatados casos de fetotoxicidade, atrasos de crescimento intrauterino e canal arterial patente. Estas anomalias no desenvolvimento estão parcialmente imputadas a uma ação direta dos inibidores da ECA no sistema renina angiotensina fetal e parcialmente à isquemia resultante de hipotensão materna e diminuição no fluxo sanguíneo feto-placentário e aporte de oxigénio/nutrientes para o feto.
Lercanidipina:
Os dados não clínicos não revelaram riscos especiais para os seres humanos, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade, potencial carcinogénico e toxicidade reprodutiva.
Os efeitos relevantes que foram observados em estudos a longo termo em ratos e cães estavam relacionados, direta ou indiretamente, com os efeitos conhecidos de doses elevadas dos antagonistas do cálcio, refletindo predominantemente a atividade farmacodinâmica exagerada.
O tratamento com lercanidipina não teve efeitos na fertilidade ou capacidade reprodutora geral, em ratos, mas doses elevadas induziram perdas pré e pós implantação e atrasos no desenvolvimento fetal. Não houve evidência de quaisquer efeitos teratogénicos em ratos e coelhos, mas outras dihidropiridinas mostraram-se teratogénicas em animais. A lercanidipina induziu distocia quando administrada em dose elevada (12 mg/kg/dia) durante o parto.
A distribuição de lercanidipina e/ou os seus metabolitos em animais em gestação e a sua excreção no leite materno não foram investigadas.
Núcleo:
Lactose mono-hidratada
Celulose microcristalina
Carboximetilamido sódico (Tipo A)
Povidona K 30
Bicarbonato de sódio
Estearato de magnésio
Revestimento (Opadry Laranja 02F23516):
Hipromelose 5cps
Dióxido de titânio (E171)
Macrogol 6000
Óxido de ferro amarelo (E172)
Talco
Óxido de ferro vermelho (E172)
Não aplicável.
2 anos.
Conservar na embalagem de origem para proteger da luz e humidade. Não conservar acima de 25ºC.
Blister de PA/Alu/PVC-Alu:
Embalagens de 7,14,28,30,35,42,50,56,90,98 e 100 comprimidos. É possível que não sejam comercializadas todas as apresentações.
Qualquer medicamento não utilizado ou resíduos devem ser eliminados de acordo com as exigências locais.
Jaba Recordati, S.A.
Av. Jacques DelorsEd. Inovação 1.2, Piso 0 - Taguspark
2740-122 Porto Salvo
Portugal
Telefone: +351 214329500
Fax: +351 219151930
Nº registo: 5605977 - 14 comprimidos
Nº registo: 5606009 - 56 comprimidos
Data da primeira autorização: 22 Maio 2014.
